











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El Lupus Eritematoso Sistémico (LES) se distingue por una diversidad de cambios en la capacidad del sistema inmunológico para tolerar sustancias propias del cuerpo, lo que resulta en una inflamación generalizada debido a la ausencia de regulación adecuada de las respuestas del sistema inmunitario. Esta condición se manifiesta con una amplia gama de síntomas clínicos, que van desde lesiones cutáneas leves hasta disfunción de órganos. Dada la complejidad del LES y la insuficiencia de tratamientos efectivos disponibles, se considera una de las enfermedades autoinmunes más desafiantes1.
El término "lupus" (que significa "lobo" en latín) fue acuñado por Hildricus, quien lo asoció con la apariencia de las lesiones cutáneas faciales, sugiriendo una similitud con las mordeduras de lobos y el adjetivo "eritematoso" fue introducido por Cazenave en 1851 para describir el enrojecimiento característico o eritema en forma de alas de mariposa en la región malar. Fue entre 1872 y 1895 que William Osler utilizó por primera vez los términos "diseminado" o "sistémico" para referirse a esta enfermedad, y posteriormente, en 1954, Harvey eliminó el término "diseminado", estableciendo así el nombre fundamental de "lupus eritematoso sistémico"2.
El LES afecta predominantemente a mujeres de entre 21 y 40 años, con una incidencia más alta entre las poblaciones de raza negra, asiática e hispana, según estudios epidemiológicos ajustados por edad y sexo. La variación en la epidemiología del LES a nivel mundial se atribuye a diferencias genéticas, demográficas, ambientales y socioeconómicas. Por ejemplo, estudios como el Proyecto de Vigilancia del Lupus de California han señalado una incidencia y prevalencia significativamente mayores entre las mujeres negras3-7.
Los interferones (IFN) son potentes citoquinas antivirales que modulan la inmunidad en respuesta a infecciones u otras señales de peligro8-9. Se han identificado más de 20 formas de IFN, y éstas se han clasificado en tres familias (tipo I, tipo II y tipo III), en función de sus distintas estructuras, ligando a receptores y actividades biológicas (1). La familia de IFN tipo I es la más grande y comprende cinco clases (IFN-α, IFN-β, IFN-ω, IFN-κ e IFN-ε), de las cuales hay 12 subgrupos adicionales de proteínas IFN-α, que codifican 13 genes altamente homólogos agrupados en el cromosoma 910-15. Además de sus funciones antivirales, los interferones son importantes en la patogénesis de las enfermedades autoinmunes, los más estudiados han sido del tipo I (IFNα e IFNβ) (9-12,16-18). La familia de IFN tipo II tiene un solo miembro, IFN-γ, que media funciones proinflamatorias e inmunomoduladoras1,17. Los interferones de tipo III (IFNλs) se describieron inicialmente como un sistema especializado que inhibe la replicación viral en las superficies de barrera epiteliales; pero recientemente se ha descrito como un importante mediador de la respuesta inmunitaria, tanto innata como adaptativas y también podrían ser responsables en EA sistémicas12,16,18-20.
El interferón de tipo I y tipo III (IFN-λ), son producidos principalmente por células dendríticas plasmacitoides (CDp). Sin embargo, la producción de IFN-λ es más abundante en las células epiteliales de la mucosa en respuesta a una infección viral, teniendo un mayor potencial citotóxico que los IFN tipo I y los IFN tipo II12,17,21,22.
Cabe señalar que el IFN tipo III es estructuralmente diferente del tipo I, sin embargo, se superponen en las acciones y ambos activan la vía del transductor de señal JAK/STAT para producir la transcripción de genes estimulados por IFN (ISG). Una diferencia importante entre los interferones tipo I y tipo III es la expresión de sus respectivos complejos de receptores. El receptor de IFNα (IFNAR) se expresa ampliamente en casi todos los tipos de células del cuerpo, mientras que la expresión del receptor IFN-λ (IFNLR) es más limitada. Además, IFN-λ tiene efectos más especializados en los sitios de barreras anatómicas(12, 16,17,23).
En estudios de expresión génica usando micromatrices, la regulación positiva de ISG (cualquier gen inducido durante una respuesta de IFN) intervienen en más del 50% de la patogenia en pacientes con LES, identificando más de 90 loci de riesgo, estableciendo varias vías críticas involucradas en su patogénesis, incluidas las respuestas inmunitarias e innatas, activación de linfocitos y formación de inmunocomplejos10,11,24.
El objetivo de la presente revisión consiste en plasmar la evidencia científica actual relacionada con el papel del IFN-λ en la patogenia del LES, centrándose en sus vías de señalización.
MATERIALES Y MÉTODOS
Se realizó búsqueda sistémica en las bases de datos, Google Académico, Nature, PubMed, The Cochrane Library y Science Direct. Se realizó la búsqueda en un período comprendido entre el 01 de enero de 2010 hasta el 10 de septiembre de 2022, para documentar la última evidencia científica. A través de las palabras claves del MeSH and DeCS: SLE; Interferons; Interferon Receptors; Autoimmunity; Toll-Like Receptors. Fue incluido adicionalmente en los criterios de búsqueda la palabra “Lambda Interferon”. Además, se decidió adicionar 7 artículos publicados anteriores a esta fecha, que consideramos significativos para explicar el objetivo de la revisión teniendo en cuenta su nivel de evidencia científica y que a la fecha no han tenido cambios. Se incluyeron artículos experimentales y originales; teniendo en cuenta criterios como tipo y clasificación de la revista y calidad de la publicación. Se excluyeron cartas al editor o sin acceso al texto completo. Posterior a la identificación de la literatura, rastreo, elegibilidad e inclusión de los registros obtenidos en la búsqueda bibliográfica, se procedió a la concepción y diseño de la propuesta, análisis de la información, redacción del manuscrito y aprobación de su versión final.
RESULTADOS Y DISCUSIÓN
Generalidades sobre IFN-λ
Existen hasta la fecha cuatro miembros de la familia tipo III (IFN-λ1-4) ubicados en el cromosoma 19, denominados también: interleucina-29 (IL-29; IFN-λ1), IL-28A (IFN-λ2), IL-28B (IFN-λ3) e IFN-λ4. Este último no está expresado en todos los humanos (1,12, 25,26). Un polimorfismo de dinucleótido común en el locus de IFNλ, puede resultar en una variante de cambio de marco que crea un nuevo gen, denominado IFN-λ4, que codifica la proteína interferón-λ4, que es moderadamente similar a IFN-λ3, que induce la fosforilación de STAT1 y STAT2 y la expresión de ISG27.
IFN-λ1-4 están estructuralmente relacionados con la familia IL-10 e interactúan con receptores heterodímeros específicos (una cadena de unión a ligando específica de IFN-λ (IFNλR1) y una cadena compartida de IL-10Rβ (subunidad además de los receptores para IL-10, IL-22 e IL-26)28. Además, comparten similitudes funcionales con los IFN de tipo I, aunque IFN-λ es estructuralmente distinto, ambos señalizan a través de la vía del transductor de señal y activador de la transcripción (STAT) (STAT1, STAT2) de la Janus quinasa (JAK) (JAK1, TYK2) para inducir la transcripción de ISG y promover la actividad antiviral1,29.
IFN-λ es importante en la inmunidad de las barreras epiteliales (células epiteliales del tracto respiratorio, intestinal y reproductivo, hepatocitos y queratinocitos). Son secretados principalmente por monocitos, CDp, queratinocitos y células epiteliales bronquiales como respuesta a una infección viral30,31). También se expresa en los macrófagos, lo que da como resultado una mejora funcional mediada por IFN-λ al mismo tiempo que promueve su secreción de quimiocinas (CXCL10 (IP-10), CCL2 (MCP-1) y CCL19 (MIP-3B) y citocinas (TNF, IL1B, IL-12 e IL-18) para la función de las células NK (citotoxicidad) y la producción de IFNγ (25). Las células B humanas expresan IFNλR su estimulación promueve la expresión de ISG, generando producción de anticuerpos mediada por el receptor tipo Toll 7 (TLR7) y TLR8 y su diferenciación a plasmablastos (12). Algunos estudios sugieren que las células T (CD4 y CD8) podrían potencialmente adquirir capacidad de respuesta a IFNλ, por inducción de ISG32).
Funciones del IFN-λ en las células del sistema inmunitario.
Las CD estimuladas con IFN-λ pueden aumentar la expresión de moléculas del complejo mayor de histocompatibilidad (MHC) de clase I y II, y niveles de moléculas coestimuladoras, que promueven la activación de las células T. Además, induce la proliferación de células T reguladoras CD4+ CD25+ Foxp3+ dependiente de IL-233.
La expresión de IFNLR en las células T es mínimo, algunos informes relacionan los IFN-λ con la desviación de las células T, hacia un fenotipo Th132. Además, estudios recientes han demostrado que las células T CD4 pueden expresar el receptor específico de IFN-λ1, el cual reduce las respuestas Th2 existentes al suprimir las citoquinas (IL-4, IL-5, IL-13) y a su vez, aumenta las diferentes citocinas Th1 (IFN-γ, TNF). Por lo tanto, con la producción de estas citoquinas, los IFN-λ favorecen la generación de DC tolerogénicas que van a estimular al alza las funciones del IFN tipo I34-36.
La señalización de IFN-λ en las células B, ha sugerido que el IFN-λ3 puede causar un aumento en la producción de IgG y la activación de las células B (12,36,37). En un estudio realizado por Goel RR, et al (17). demostraron que la supresión genética de IFNLR1 protege a los ratones de la desregulación inmunitaria y del daño orgánico en un modelo de Lupus inducido por TLR7, actuando, así como regulador para inhibir las respuestas inmunitarias de las células B y la producción de IgG21,37).
Los neutrófilos expresan altos niveles de IFN-λR1; pero en estas células a diferencia de los linfocitos T y B, IFN-λ inhiben su reclutamiento y activación, evitando la amplificación de la inflamación38. Estudios en neutrófilos de ratón muestran que los IFN-λ activan JAK2 e inhiben la producción de especies reactivas de oxígeno (ROS) en un modelo de inflamación intestinal. Este efecto estuvo mediado por la inactivación de RAC-alfa serina/treonina-proteína quinasa (AKT) mediada por JAK2 y este efecto dependía de la capacidad única de IFN-λ (no los interferones de tipo I) para activar JAK239.
Lo anterior sugiere que los IFN-λ pueden regular al alza o a la baja las respuestas celulares en entornos inmunoespecíficos. Sin embargo, se han encontrado resultados inconsistentes en diferentes modelos29).
Señalización por IFN-λ
Los receptores de reconocimiento de patrones ubicados en el endosoma (TLR3/7/8/9), en la membrana celular (TLR4) o el citoplasma (secuencia activada por IFNγ (cGAS), proteína 5 asociada a la diferenciación de melanoma (MDA-5), detectan ácidos nucleicos virales para desencadenar una vía de señalización que da como resultado la producción de interferones tipo I y III. Los interferones tipo I y tipo III pueden activar tanto la JAK1 como la tirosina cinasa no receptora (TYK2), conduciendo a la fosforilación del transductor de señales y activación de la transcripción STAT, formando heterodímeros STAT1-STAT2. Estos heterodímeros interactúan con el factor regulador de interferón 9 (IRF9) para formar el complejo de transcripción del factor 3 del gen estimulado por interferón (ISGF3). ISGF3 se transloca al núcleo, donde puede unirse a secuencias de elementos reguladores estimulados por interferón (ISRE) y promover la expresión de ISG (IFI6, USP18, ZCCHC2, SERPING1, SP100, SAMD9, RSAD2, PHF11, HERC5, MX1, USP18, OAS2, LY6E, IFI27, PLSCR1)40. Los interferones tipo I y tipo III también pueden promover la formación de homodímeros STAT1, que regulan al alza la expresión del factor regulador de interferón 1 (IRF1) y conducen a la producción de quimiocinas proinflamatorias (CXCL9,CXCL10, CXCL11). IFN-λ también puede señalar a través de una variedad de mecanismos no canónicos17 (Figura 1).
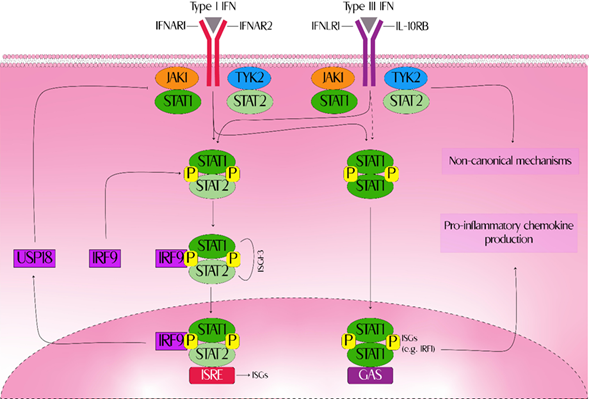
IFN-λ1-4 están estructuralmente relacionados con la familia IL-10 e interactúan con receptores heterodímeros específicos (cadena de unión a ligando específica de IFN-λ (IFNλR1) y cadena compartida de IL-10Rβ (una subunidad de los receptores para IL-10, IL-22 e IL-26), denominándose como receptor. Además, comparten similitudes funcionales con los IFN de tipo I, aunque IFN-λ es estructuralmente distinto, ambos señalizan a través de la vía del transductor de señal y activador de la transcripción (STAT) (STAT1, STAT2) de la Janus quinasa (JAK) (JAK1, TYK2) para inducir la transcripción de genes estimulados por interferón (ISG) y promover la actividad antiviral. Los interferones tipo I y tipo III pueden activar JAK1 como TYK2, conduciendo a la fosforilación del transductor de señales y activación de la transcripción (STAT) formado heterodímeros STAT1-STAT2. Estos heterodímeros interactúan con IRF9, para formar el ISGF3. ISGF3 se transloca al núcleo, donde puede unirse a ISRE y promover la expresión de ISG. Los interferones tipo I y tipo III también pueden promover la formación de homodímeros STAT1, que regulan al alza la expresión de IRF1 y conducen a la producción de quimiocinas proinflamatorias. IFN-λ también puede señalar a través de una variedad de mecanismos no canónicos. Modificado de 13. Elaboración: propia.
GAS: secuencia activada por IFNγ; IFN: interferón; IFNAR: receptor de IFNα; IFNLR1: receptor 1 de IFNλ; IL-10RB: subunidad β del receptor de IL-10; IRF1: factor regulador de interferón 1; IRF9: factor regulador de interferón 9; ISG: genes estimulados por interferón; ISGF3: factor 3 del gen estimulado por interferón; ISRE: secuencias de elementos reguladores estimulados por interferón JAK1: Janus cinasa 1; TYK2: tirosina cinasa no receptora; USP18: peptidasa 18 específica de ubiquitina.
Es importante destacar que el sensor citosólico Ku70 parece estar preferentemente involucrado en el reconocimiento de IFN-λ en lugar de IFN tipo I1,30 (Figura 2).
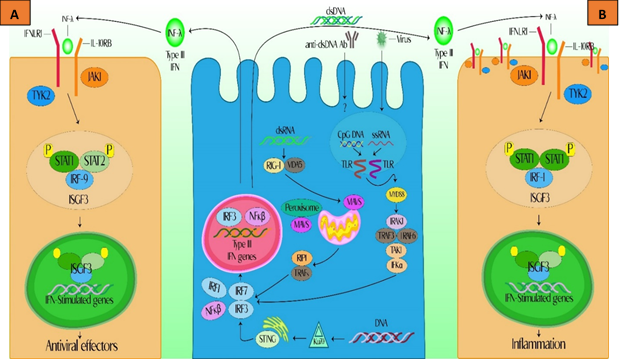
A.) Los IFN tipo III pueden ser inducidos por una infección viral o por inmunocomplejos, que son detectados por los PRR, especialmente TLR y RIG-I y RLR. Las moléculas de señalización diferencial conducen a la activación de NF-κB y IRF, finalmente, a la activación de las transcripciones del gen IFN. Los IFN secretados se unen a los receptores de IFN tipo III (IFNLR)1/receptor de interleucina-10 (IL-10R)2, de células vecinas y estimulan la producción de ISG a través de las vías JAK/STAT, lo que da como resultado la producción de varios efectores antivirales. La GMP-AMP sintasa cíclica (cGAS) transforma el ADN en dinucleótidos cíclicos (CDN), que pueden ser reconocidos por el estimulador de genes de interferón (STING). STING desencadena la activación de la quinasa 1 de unión al tanque (TBK1) para fosforilar IRF-3, involucrada en Ku70 e induce la transcripción de IFN de tipo III. B.) La inducción de IRF-1 depende de la expresión de IFNLR-1, ya que la sobreexpresión de IFNLR-1 aumenta la cantidad de quimiocinas CXC producidas en respuesta a IFN-λ a niveles similares a los provocados por IFN de tipo I. Estos hallazgos sugieren que la cantidad de IFNLR-1 es un determinante importante de la función de IFNλ. Modificado de 1,25. Elaboración: propia.
DNA: ácido desoxirribonucleico; dsRNA: RNA de doble cadena; NF-κB: factor nuclear potenciador de la cadena ligera kappa de las células B activadas; FNLR1: receptor 1 de IFNλ; IL-10R: receptor de IL-10; IRAK1: Quinasa 1 asociada al receptor de interleucina-1; IRF: factores reguladores del interferón; ISG: genes estimulados por IFN; ISGF3: factor 3 del gen estimulado por interferon; JAK: Janus quinasa; MAVS: adaptador mitocondrial de señalización antiviral; MDA5: proteína 5 asociada a la diferenciación de melanoma; MYD88: respuesta primaria de diferenciación mieloide 88 ; RIP1: Proteína ligada al receptor 1; PRR: receptores de reconocimiento de patrones; RIG-I: receptores similares al gen 1 inducible por ácido retinoico; STAT: Transductor de Señal y Activador de Transcripción; TAK1: adaptador de la quinasa 1; STING: estimulador de genes de interferón; TRAFs: Factores asociados al receptor del factor de necrosis tumoral; TLR: receptores tipo Toll; TYK2: tirosina cinasa no receptora.
Fisiopatología del Lupus e IFNλ
En pacientes con LES, la producción aberrante de interferón tipo I es desencadenada por los ácidos nucleicos propios, originando inmunocomplejos autorreactivos, que a menudo se generan como subproducto de una deficiencia en la eliminación de células apoptóticas o por Netosis. En combinación con una pérdida de la señal de retroalimentación negativa, así como mutaciones en ISG. En respuesta a estos desencadenantes, existe una producción crónica de interferón tipo I en los sistemas inmunitarios innato y adaptativo, promoviendo la formación de homodímeros STAT1 que se unen al promotor IRF1, induciendo la expresión de IRF1. Este proceso induce la producción de más autoanticuerpos e interferón tipo I. Teniendo en cuenta que la vía de activación para interferón tipo I y tipo III es muy similar, se considera que una producción aberrante de interferón tipo I produce la desregulación la alza de IFN-λ, a través de ISG, interviniendo así en la fisiopatología del LES1,6,9,12,18,25,37).
En pacientes con LES, los depósitos de complejos inmunes y la inflamación crónica subsiguiente en los tejidos contribuyen al daño orgánico irreversible. La CDp desempeñan un papel predominante en el bucle de autoamplificación que impulsa la producción de interferón tipo I, generando depósitos de inmunocomplejos, inflamación crónica tisular constante, contribuyen a la maduración de linfocitos B mediante la presentación de antígeno, y generación de autoanticuerpos20,41. Además, el IFN-λ puede ser inducido directamente por el gen estimulador de IFN tipo 3 (ISGF3) u otros factores de transcripción inducidos por IFN, como IRF-11,16. Así mismo, IFN-λ no inducen suficiente expresión de IRF-1 para permitir la producción de quimiocinas. En particular, la inducción de IRF-1 depende de la expresión de IFNLR-1, ya que la sobreexpresión de IFNLR-1 aumenta la cantidad de quimiocinas CXC producidas en respuesta a IFN-λ a niveles similares a los provocados por IFN de tipo I. Estos hallazgos sugieren que la cantidad de IFNLR-1 es un determinante importante de la función de IFN-λ. Concluyendo teóricamente que podrían promover la inflamación si la expresión de IFNLR-1 es lo suficientemente alta como para inducir la expresión de IRF-1(17, 42,43) (Figura 2).
Estudios en los hepatocitos humanos sugieren que IFNα puede aumentar la expresión de IFNLR1 y que este efecto depende del genotipo de IFN-λ3 (rs12979860 y rs8099917)44.
Podemos decir que, en el LES, los autoanticuerpos que forman inmunocomplejos detectan los ácidos nucleicos propios de las células apoptóticas y las trampas extracelulares de neutrófilos (NET), lo que estimula a las CDp para que produzcan IFN tipo I y tipo III. Además, las células epiteliales producen IFN de tipo III. Tanto los IFN tipo I como III estimulan las CD y, por lo tanto, conducen a una activación inapropiada de las células T y B, que contribuyen a una mayor producción de citocinas proinflamatorias y autoanticuerpos16,20,41.
Se demostró que la expresión de IFNλR-1 aumentaba en células B de memoria y vírgenes, y la estimulación de células B vírgenes por el receptor de células B (BCR) y por IFN-λ, indujo una mayor diferenciación a células plasmáticas. Asimismo, IFN-λ junto con la estimulación de TLR7/8 de células B aumentó la expresión del marcador de activación temprana CD69, IL-6 e IL-10, generando producción de anticuerpos y proliferación. Esta activación de las células B podría desempeñar un papel crucial en la fisiopatología del LES12. En un estudio realizado por Goel et al.12, afirmaron que los IFN tipo III tienen un papel fundamental en la inflamación lúpica asociada a TLR7. Promoviendo la no regulación inmunitaria a través de efectos inflamatorios localizados en la piel y los riñones. Concluyendo que genera mayor nefritis lúpica porque las células renales expresan el receptor de IFN-λ3 y podrían ser muy susceptibles a la apoptosis inducida por IFN-λ, lo que provoca inflamación, necrosis y daño renal. Por su parte los queratinocitos responden directamente al IFN-λ, produciendo moléculas proinflamatorias que se unen al receptor de quimiocinas CXCR3 y promueve el reclutamiento de monocitos y linfocitos con expresión aumentada de MHC-I, involucrado en potenciar la respuesta de células T CD8+, a los sitios de inflamación por quimiocinas asociadas (CXCL9, CXCL10, CXCL11).
IFN-λ podrían tener efectos sobre la tolerancia central y la selección de células T en el timo. IFN-λ se expresa constitutivamente en células epiteliales medulares tímicas (TMEC) y promueven la expresión de moléculas MHC de clase I en las TMEC. La expresión de MHC de clase I inducida por IFN-λ parece ser crucial para la selección efectiva de células T45.
Por lo tanto, la señalización de IFN desregulada ahora se reconoce como una nueva familia de enfermedades denominadas "interferonopatías"46. Estudios recientes han sugerido que quizás el 10% de nuestros genes están regulados por IFN, pero el IFN de tipo III induce un número limitado de genes y no se han definido transcritos únicos47. Los pacientes con LES muestran un patrón más complejo de expresión génica16.
Correlación clínica entre IFN tipo III y LES (signos y síntomas de la enfermedad)
En un estudio de casos y controles realizado por Abdelraouf et al.22, en el año 2022, donde evaluaron los niveles séricos de IFN-λ3 en 40 pacientes (35 mujeres y 5 hombres) egipcios con LES y 40 pacientes controles, investigaron su potencial relación con el índice de actividad de la enfermedad del LES (SLEDAI). Obteniendo como resultado que los niveles séricos de IFN-λ3 fueron más altos en pacientes con LES (9,7 ± 12,47 pg/mL), en comparación con el control (5,13 ± 1,63 pg/mL) (p = 0,02). Se observaron correlaciones significativas entre IFN-λ3 sérico y serositis (r= 0,35, p= 0,03), consumo de la fracción del complemento 3 (C3, r −0,33, p= 0,04) y SLEDAI (r 0,34, p= 0,03). En el análisis de regresión multivariable, la serositis y SLEDAI (pero no C3) fueron predictores significativamente independientes de los niveles de IFN-λ322. Estos resultados fueron consistentes con los de Amezcua-Guerra et al., quienes investigaron los niveles séricos de IFN-λ e IFN-α en pacientes con LES en México y reportaron un nivel significativamente mayor de IFN-λ3, pero no de IFN-λ1, IFN-λ2 e IFN-α18).
En un metaanálisis realizado por Wang et al., en 2022, incluyeron 30.604 participantes de origen europeo, chino y tailandés. Utilizando datos epigenómicos públicos y loci de rasgos cuantitativos de expresión. Encontraron que una deleción de 1pb (pares de base) corriente arriba del gen para IFNLR-1 estaba asociada con LES. Lo que proporciona evidencia de un papel de la señalización de IFN-λ en el LES47.
Otro estudio observacional realizado por Munes et al.48, quería determinar si un polimorfismo en el gen IL28RA se asocia con AR y LES y subfenotipos de enfermedades específicas. Investigaron en 603 individuos brasileños (178 LES y 176 AR) y 249 controles. La variante IL28RA (rs4649203) se genotipificó mediante el ensayo TaqMan. Encontrando que el alelo rs4649203-G (menor) se asoció con la aparición de LES y AR y se demostró que es un factor de riesgo de serositis y anemia entre los pacientes con LES, así como un factor protector de vasculitis reumatoide y nódulos reumatoides en pacientes con AR, lo que sugiere una asociación con una forma más leve de la enfermedad.
El estudio realizado por Chen et al. Donde utilizaron ensayos de discriminación de alelos TaqMan para determinar los polimorfismos de un solo nucleótido (SNP) IFN-λ3/4 en 1.620 controles (701 hombres y 919 mujeres) y 1.013 (71 hombres y 760 mujeres) pacientes con LES de Taiwán, comparándolo entre pacientes con LES y controles y entre pacientes con LES estratificados por fenotipos clínicos. Concluyeron que todos los alelos principales de SNP IFN3/4 se asociaron significativamente con el riesgo de LES. Además, todos los SNP IFN3/4 de alelos menores se asociaron significativamente con la susceptibilidad a nefritis lúpica en comparación con los controles. También los niveles séricos elevados de IFN-λ3 se correlacionaron significativamente con la disminución del complemento (C3-C4) y la actividad elevada de la enfermedad49. Por el contrario, otro estudio realizado por Juárez et al.29, quisieron comparar si el SNP rs12979860 en el IFN-λ3/4 que se asoció significativamente con la susceptibilidad al LES en pacientes taiwaneses, también estaba asociado con la presencia de LES y nefritis lúpica en individuos mexicanos, así como con la expresión de varios ISG en pacientes con LES. En total, genotiparon 439 pacientes con LES y 358 controles para rs12979860 mediante PCR en tiempo real y se construyeron gráficos de discriminación alélica. El análisis de casos y controles reveló que rs12979860 no se asoció con la susceptibilidad al LES (OR 1,18, IC del 95 % 0,97-1,45, p = 0,08) ni con el riesgo de nefritis lúpica (OR 0,913, IC del 95 % 0,590-1,411, p= 0,682). Esta diferencia en los resultados con la investigación de Chen et al, pudo explicarse por poder estadístico inadecuado debido al tamaño de muestra limitado y/o las diferencias raciales y étnicas49.
Un nuevo estudio realizado por Oke et al.50, donde participaron 97 pacientes con LES y 322 controles, midieron la actividad de la enfermedad SLEDAI y la medida de actividad del lupus sistémico (SLAM), comparándolas con los niveles de IFN-α, IFN-λ1 e IFN-γ. Concluyendo que todas las mediciones de IFN fueron más altas en los pacientes con LES. La actividad alta de IFN tipo I se correlacionó con los niveles de IFN-γ e IFN-α y se asoció con LES activo en la mayoría de los dominios: pérdida de peso, fatiga, fiebre, erupción cutánea, linfadenopatía, artritis, nefritis y manifestaciones hematológicas. Los subconjuntos específicos de LES se vincularon con la regulación positiva de diferentes subtipos de IFN circulantes: IFN-γ alto para artritis, nefritis y anticuerpos anti-Ro e IFN-α alto para compromiso mucocutáneo y anticuerpos anti-Ro y anti-La. El IFN-λ1 alto aislado se asoció a anticuerpos antinucleosoma y LES menos grave; por lo que la afectación de diferentes órganos parece estar asociada a diferentes tipos de IFN. Se ha observado una mayor expresión de IFN-λ1 e IFNκ en la piel de pacientes con Lupus cutáneo, y en nefritis por Lupus; las biopsias renales muestran una mayor expresión de genes inducibles por IFN-λ y las CDp se acumulan en los glomérulos de pacientes con enfermedad activa16. La asociación con la serositis podría deberse a la abundancia de IFN-λ en las superficies epiteliales. Además, el aumento del IFN-λ aumenta los niveles de anti-dsDNA (51), y disminuyen los niveles del complemento52.
CONCLUSIÓN
En el LES la producción de IFN-λ, promueve activación de células del sistema inmune, generando depósitos de inmunocomplejos, inflamación crónica tisular y autoanticuerpos.
En particular, los niveles séricos elevados de IFN-λ3 se correlacionan significativamente con una mayor actividad de la enfermedad, además parece ser factor de riesgo para producir nefritis lúpica. Se sugiere que IFN-λ3 podría servir como un biomarcador potencial para el monitoreo de la actividad de la enfermedad y predecir pronóstico en LES.
Se necesitan más investigaciones futuras para un mejor conocimiento de las vías de activación y las actividades superpuestas de los diferentes IFN.

