











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los tumores neuroendocrinos (NET) derivan de las células enterocromafines del tracto gastrointestinal y fueron descritos por Lubarsch en 1.8881. Desde 1.974, los tumores neuroendocrinos (NETs) se han clasificado de acuerdo con las principales hormonas secretadas, tales como insulinoma, gastrinoma, VIPoma, glucagonoma y somatostatinoma.
Desde los dos primeros casos de somatostatinoma informados en 1.977, menos de 200 casos de somatostatinoma han sido reportados2. El somatostatinoma es un tumor raro que se localiza en páncreas o duodeno con una incidencia de 1:40 millones, cuya presentación clínica es variable3. En los pacientes con neurofibromatosis tipo I la incidencia de este tipo de tumor es de alrededor al 1% y suelen asociarse con adenocarcinomas, tumores de GIST y feocromocitoma. Aquellos de localización duodenal presentan con menor frecuencia el síndrome del somatostatinoma caracterizado por diabetes mellitus, colelitiasis y esteatorrea convirtiendo en más difícil si cabe su diagnóstico preoperatorio5. Las manifestaciones clínicas más frecuentes en los no-funcionantes son: la ictericia (65%), dolor abdominal inespecífico (31%) junto con hemorragia digestiva baja, anemia ferropénica, oclusión intestinal, colangitis y pancreatitis5. En somatostatinomas duodenales, el síndrome del somatostatinoma puede ocurrir sólo si el tumor es mayor de 4 cm6. Debido al potencial maligno de estos tumores el gold standard del tratamiento es la cirugía, considerándose la resección local o endoscópica de las lesiones menores de 1 cm7. El riesgo de metástasis aumenta significativamente en tumores de 2 cm. Para tumores más grandes (> 2 cm), la cirugía de Whipple con linfadenectomía regional debe ser considerada7.El tratamiento adyuvante con análogos de la somatostatinaha demostrado un aumento en la supervivencia. La sobrevida a los 5 años de los somatostatinomas con metástasis hepáticas es del 40%, sin embargo, en los tumores sin metástasis hepática o ganglionar es del 100%6.
CASO CLÍNICO
Paciente de sexo masculino de 47 años de edad, portador de Enfermedad de Von Reckling hausen, sin tratamiento. Refiere cuadro de 1 mes de evolución de vómitos en varias oportunidades, de contenido alimentario, precedido de náuseas. Se agrega al cuadro pérdida de peso de 15 kg, coloración amarillenta de piel y mucosas, astenia y anorexia.
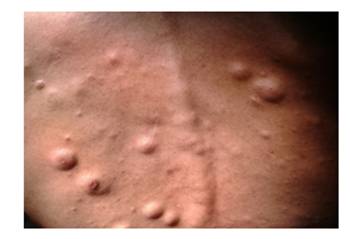
Examen físico: Piel y mucosas: neurofibromas y manchas café con leche en miembros superiores e inferiores, tórax y abdomen (Figura 1). Resto sin datos de valor.
Estudios laboratoriales: Hb. 12,7g/dl Hto. 37,9% GB: 12.300 N 80% B.T: 2,90 B.D: 1,3 F.A: 570 G.O.T: 57 G.P.T: 90 Amilasa: 1065 Lipasa: 2054 CEA 125 7,39 U/ml CA19-9 25,2 U/m
Ecografía abdominal: vías biliares intrahepáticas dilatadas, colédoco dilatado, mide 16 mm de diámetro, vesícula biliar: aumentada de volumen, pared no engrosada, contenido barro biliar. Páncreas no visualizado por interposición de gases. Se constata una imagen heterogénea en flanco derecho. En TAC de abdomen se observa dilatación del conducto biliar y pancreático y un tumor de 3,5 cm en región periampular (Figura 2). Se realiza EDA que informa en la 2da. porción del duodeno lesión de aspecto adenomatoso que ocupa casi toda la circunferencia. Se recibe informe de biopsias: en la lámina propia un adenocarcinoma moderadamente diferenciado. Se indica cirugía, realizándose una duodeno pancreatectomía cefálica (Figura 3), la anatomía patológica informa tumor neuroendocrino bien diferenciado de duodeno. El tumor se ajusta a la clasificación de Somatostatinoma duodenal o periampular T3 N1 Mx Estadio IIIB.
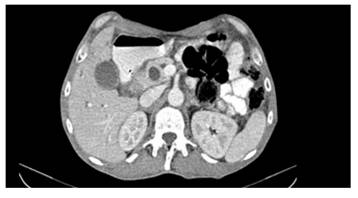
Figura 2. La tomografía computarizada de abdomen muestra una masa bien definida en la segunda porción del duodeno.

DISCUSIÓN
En pacientes con neurofibromatosis tipo I la incidencia de este tipo de tumores es del 1 %. El gold standard del tratamiento es la cirugía.
En el paciente se realizó la cirugía de Whipple, teniendo en cuenta que el tamaño del tumor era mayor a 2 cm. Durante el post operatorio presentó una fístula quilosa que fue tratada con dieta hipograsa, hiperproteica, NPT y análogos de la somatostatina. Fue dado de alta al 30mo. día post operatorio.
El somatostatinoma debe sospecharse en pacientes afectos de neurofibromatosis tipo I cuya optimización del diagnóstico y tratamiento representan un reto terapéutico.

