











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
En el manejo de las convulsiones neonatales, resulta de mucha importancia determinar la etiología de las mismas, ya que permite instaurar una terapia adecuada y limitar la disfunción del sistema nervioso central, que ocurriría si éstas no fueran tratadas correctamente. Como existen afecciones que precisan de una terapia específica para lograr el control de las convulsiones, es imprescindible aclarar la etiología1.
Si bien existe discusión con respecto al efecto de las convulsiones sobre el cerebro inmaduro, actualmente se sabe que el resultado a largo plazo depende de la etiología de la convulsión, y el grado o severidad de la injuria cerebral causada por el disturbio subyacente1.
Aunque la mayoría de las convulsiones neonatales son manifestaciones agudas de otros procesos; sin embargo hay un creciente reconocimiento de la existencia de síndromes epilépticos de comienzo neonatal. Entre ellos se citan los siguientes: convulsiones neonatales benignas, epilepsia neonatal familiar benigna, encefalopatía mioclónica temprana, encefalopatía epiléptica infantil temprana, encefalopatía KCNQ2 y el síndrome DEND (retraso mental, epilepsia, y diabetes neonatal)2.
Se considera síndrome epiléptico neonatal severo, a los casos en los que no se halla una causa específica para las convulsiones, es decir éstas no son síntomas de una afección, y el electroencefalograma interictal muestra un patrón de ondas-supresión; además las convulsiones son refractarias al tratamiento, y se asocian a pobre resultado en el neuro desarrollo1.
Dentro de este grupo de síndromes epilépticos severos se incluyen la encefalopatía mioclónica temprana, y la encefalopatía epiléptica infantil temprana o síndrome de Ohtahara.
Las enfermedades epilépticas no son afecciones raras; alrededor de un 40% de las convulsiones que se presentan en los primeros años de vida son debidas a encefalopatía epiléptica3. Por todo lo mencionado con anterioridad, consideramos importante que los pediatras generales también estén informados sobre aspectos clínicos, diagnóstico y manejo terapéutico de estas afecciones; por ello decidimos presentar este caso.
CASO CLÍNICO
RN de sexo masculino, de 15 días de vida, derivado al Hospital Nacional de Itauguá con antecedentes maternos de primípara, de 28 años, 1 aborto cuya causa era desconocida, 4 controles prenatales, sin patología aparente, con VDRL y test para VIH negativos, grupo sanguíneo A+, de 37 semanas de gestación según fecha de última menstruación y ecografía del primer trimestre. Además, rotura prematura de membranas sin especificarse la duración de la misma.
El niño nació por vía vaginal en un centro asistencial, en presentación cefálica, con doble circular de cordón y líquido amniótico claro, deprimido, con Apgar 3/5/8, requiriendo maniobras de reanimación cardiopulmonar, sin especificarse cuáles en hoja de remisión; quedando posteriormente con dificultad respiratoria, por lo que se instala oxígeno en halo cefálico y es internado. Siendo las medidas antropométricas, peso=3180 gr (Pc=75), talla=48 cm (Pc=50-75), Perímetro cefálico=32 cm (Pc=25-50), según curva de Fenton; evaluado por Capurro como de 37 semanas. Al examen físico presenta labio leporino y fisura palatina. Durante la internación recibe tratamiento con antibióticos por sospecha de sepsis neonatal con ampicilina y cefotaxima durante 10 días, no se cuenta con datos de laboratorio ni cultivos de dicho centro. Deciden el traslado por persistencia de requerimiento de oxígeno, ya que al suspenderlo desarrollaba cianosis, para descartar cardiopatía congénita.
Al ingresar el recién nacido a nuestro servicio se constató además de la presencia de nasoqueilopalatosquisis; la existencia de retrognatia, glosoptosis, micropene, e hipotonía cervical; llamando la atención la presencia de estridor laríngeo. Se intenta suspender oxígeno, sin tolerarlo, ya que sufre descensos de saturación de 75-80%.
Internado se efectúa ecocardiografía, que informa corazón estructuralmente normal, sin hipertensión pulmonar. Se asume como probable causa del requerimiento de oxígeno la obstrucción respiratoria por la glosoptosis.
Ecografìa transfontanelar informó hemorragia intraventricular grado II bilateral e imagen anecoica de 1 cm de diámetro transversal en fosa posterior, compatible con megacisterna magna, versus quiste aracnoideo (Fig. 1).
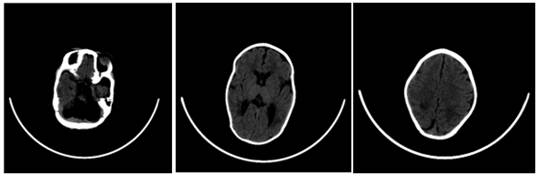
Llama la atención la probable agenesia cerebelar. Signos de paquigiria con profundización de los surcos corticales. Aparente leuco-encéfalo-malasia en región frontal y parieto-occipital bilateral con buena diferenciación cortico/subcortical. Sistema ventricular con variante que correspondería a cavum del septum pellucidum.
Se realizó interconsulta con Otorrinolaringólogo y cirujano de cabeza y cuello, planteando efectuar una glosopexia. Los especialistas agregaron al diagnóstico laringomalasia leve, y dificultad para la deglución. Sugieron manejarlo cuidando la posición del paciente, mantenerlo en posición prona. A los 14 días de internación logró suspenderse oxígeno. Se lo mantuvo con alimentación por sonda orogástrica; y se estimuló succión y deglución mediante fisioterapia, sin éxito.
A los 47 días de internación presentó crisis tónico clónicas generalizadas en miembros superiores e inferiores, con actitud en opistótonos del tronco; evaluado por neuropediatra, se sospechó síndrome de West y se indicó ácido valproico; se solicitó electroencefalograma y tomografía de cráneo simple.
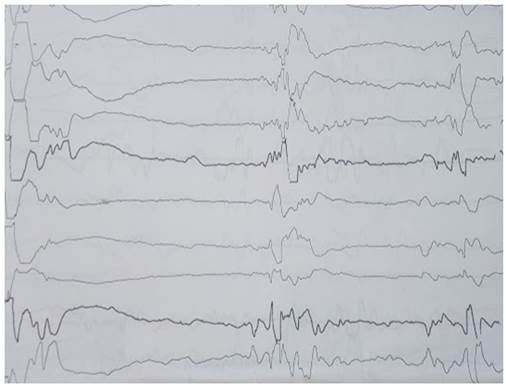
A los 63 días de vida, aumentaron las secreciones en vías respiratorias, empeoró la dinámica respiratoria, requirió nuevamente oxígeno, presentó febrícula, se sospechó infección asociada a cuidados de la salud, se lo policultivó e inició tratamiento con vancomicina más amikacina inicialmente. Se decidió iniciar el tratamiento con dichas medicaciones teniendo en cuenta el resultado de un hemocultivo tomado una semana antes, debido a episodios aislados de febrícula, en que creció estafilococo epidermidis. Después de 48 horas se decidió ampliar cobertura antibiótica con ceftazidime y claritromicina, suspendiendo amikacina. Completó 21 días de tratamiento con los antibióticos mencionados previamente, y 14 días de claritromicina. Continuó con evolución tórpida, persistencia de abundantes secreciones y requerimiento de oxígeno. En el aspecto neurológico, las convulsiones no lograron controlarse a pesar de aumentar dosis de ácido valproico, y agregar vigabatrina. A los 65 días de vida se realizó electroencefalograma que demostró un patrón de descargas-supresión; y en tomografía de cráneo se observó aparente agenesia cerebelar; signos de paquigiria con surcos cerebrales profundos y leucoencefalomalasia frontal y parietooccipital bilateral, como puede apreciarse en las imágenes de la figura 1. Evaluado por neuróloga se realizó el diagnóstico de síndrome de Ohtahara.
A los 98 días presentó de nuevo febrícula, se efectuó radiografía que mostró condensación pulmonar, se inició nuevamente tratamiento antibiótico con meropenem y vancomicina. El paciente no mostró mejoría, continuó con febrícula a pesar de 8 días de tratamiento antibiótico, aspecto pálido y reticulado de la piel. A los 110 días de vida desarrolló apnea con bradicardia extrema, luego paro cardiaco y ya no respondió a medidas de reanimación, declarándose el óbito del mismo.
DISCUSIÓN
Si bien es sabido que el efecto encefalopático de la actividad epiléptica puede ocurrir en asociación con cualquier forma de epilepsia, sin embargo, esto se presenta con mayor frecuencia en un grupo de síndromes llamados encefalopatías epilépticas de la niñez(Fig. 2). Estas entidades se caracterizan porque la actividad epiléptica por sí misma contribuye a la alteración en el desarrollo cognitivo y del comportamiento, más de lo que podría esperarse por la patología subyacente sola, como ocurre en las malformaciones corticales. Además, este deterioro clínico progresa con el tiempo; y se acompañan de una anormalidad severa en el electroencefalograma1,4.
Los siguientes síndromes son considerados encefalopatías epilépticas de la niñez: encefalopatía mioclónica temprana, síndrome de Ohtahara, epilepsia de la infancia con convulsiones focales migrantes, síndrome de West, síndrome Dravet, síndrome Doose o epilepsia con convulsiones mioclónicas atónicas, síndrome Lennox-Gastaut, encefalopatía epiléptica con continuo de picos y ondas durante el sueño, y síndrome Landau-Kleffner. Cada una de estas entidades tienen características clínicas y electroencefalográficas que permite diferenciarlas3,5.
En el año 1976 Ohtahara describió por primera vez un síndrome epiléptico que se presenta en la temprana infancia, con un característico patrón electroencefalográfico de descargas supresión, al cual denominó encefalopatía epiléptica infantil temprana. Posteriormente el mismo Ohtahara observó en el seguimiento, que los niños con esta afección evolucionaban a síndrome de West hasta en un 75% de casos y a síndrome de Lennox-Gastaut en 12%6. Este hallazgo ha conducido al origen de la teoría de que estos síndromes constituyen reacciones específicas del cerebro relacionadas a la edad, ante un mismo agente exógeno, denominándoselas encefalopatías dependientes de la edad1,7.
El caso presentado trata de un paciente con las características clínicas correspondientes al síndrome de Pierre Robin, asociado a laringomalasia y micropene. A los 62 días aparecen convulsiones difíciles de controlar, con signos de importante compromiso neurológico; hipotonía cervical desde el inicio, así como la imposibilidad de lograr la succión y deglución. Sin embargo, el hallazgo clínico no pudo atribuirse a asfixia perinatal, ya que, si bien el niño presentó depresión al nacer, no tuvo la evolución característica de pacientes con esa patología, pues las convulsiones no aparecieron dentro de las 24 horas después del nacimiento. Por otra parte, con el estudio de imágenes se descubrió la presencia de defectos anatómicos y estructurales en el sistema nervioso central. El electroencefalograma mostró un patrón de descargas-supresión; con lo cual se arribó al diagnóstico de síndrome de Ohtahara.
La encefalopatía epiléptica de inicio temprano o síndrome de Ohtahara, es una de las formas más severas de encefalopatía; de inicio precoz, dentro de los primeros tres meses de vida, y en la mayoría de los casos incluso dentro de los 10 días o las primeras horas de vida, también existen reportes de movimientos convulsivos incluso intraútero.
Las convulsiones se presentan más comúnmente como espasmos tónicos que se desarrollan en series o en forma aislada, con una duración de hasta 10 segundos; pero también pueden desarrollarse crisis parciales motoras erráticas, convulsiones tónico-clónicas unilaterales, alternantes entre ambos hemicuerpos (en báscula) y crisis generalizadas tónico-clónicas. Las crisis parciales se observan en la tercera parte de los casos4,7.
El EEG interictal muestra la presencia de oleadas generalizadas de gran amplitud (150-350 microvoltios) que alternan con fases de supresión (con una duración de 3 a 4 seg) de la actividad eléctrica cerebral (trazado de descarga-supresión). Las oleadas generalizadas están constituidas por ondas lentas polimorfas de gran amplitud mezcladas con puntas, y su duración es de 1 a 3 seg. El trazado se obtiene en estados de sueño y vigilia; y puede desarrollarse en forma sincrónica o no con los movimientos anormales. Con el tiempo las fases de supresión sufren acortamiento en su duración, predominando las descargas7,8.
El síndrome de Ohtahara puede deberse a una variedad de etiologías, siendo frecuente su asociación a alteraciones en la estructura cerebral. También se han descripto menos comúnmente casos relacionados a alteraciones metabólicas o mutaciones genéticas; algunas de éstas dos últimas etiologías pueden tener además alteraciones estructurales. Las alteraciones estructurales asociadas con el síndrome de Ohtahara incluyen: hemimegalencefalia, agenesia del cuerpo calloso, porencefalia, agenesia de cuerpos mamilares, displasia olivodentada, injuria hipóxica, displasia cortical y desórdenes de migración neuronal9-12.
Los desórdenes metabólicos hallados, relacionados al síndrome de Ohtahara son los siguientes: hiperglicemia no cetósica, deficiencia de citocromo oxidasa C, dependencia de piridoxina, deficiencia de carnitina palmitoiltransferasa, y la encefalopatía de Leigh. Recientemente se halló el síndrome de Ohtahara asociado a deficiencia de biotinidasa en un paciente; y en dos pacientes con deficiencia del complejo I de la cadena respiratoria mitocondrial. Uno de los pacientes con deficiencia del complejo I de la cadena respiratoria, evidenció microcefalia, cuerpo calloso delgado, y atrofia cortical. En tanto que el otro paciente no presentó anormalidad en estudio de imágenes9,13-15.
La deficiencia de citocromo oxidasa C y deficiencia del complejo I de la cadena respiratoria, producen un defecto en la generación de energía en las células, lo cual conduce a desmielinización y defectos de migración neuronal, paquigiria, micropoligiria13,15.
Con respecto a las mutaciones genéticas, ha habido un creciente reporte de casos de síndrome de Ohtahara en relación a mutaciones genéticas; como la mutación en el gen de la proteína de unión sintaxina, STXBP1; el gen ARX, y el gen SLC25A22. La última mutación mencionada, se suele observar en casos de consanguinidad en los padres.
El mecanismo por el cual las mutaciones genéticas actúan para ocasionar epilepsia, es a través de la disgenesia cerebral y la disfunción neuronal que producen. El gen SLC25A22 participa en el transporte de glutamato mitocondrial, su deficiencia ocasiona depleción energética que causa disfunción neuronal y muerte celular. El gen ARX participa en la regulación de la diferenciación y proliferación neuronal; además en la migración de neuronas a la corteza durante el desarrollo. La mutación en este gen se asocia a hipoplasia del cuerpo calloso, ganglios basales pequeños e hipocampo, defecto del cavum septum pellucidum, y atrofia cerebral. La disfunción en la diferenciación puede conducir a un defecto en la función de las interneuronas inhibidoras, que explican las convulsiones intratables observadas en estos pacientes.
El gen STXBP1 dirige la liberación de vesículas sinápticas, por lo tanto, como el gen ARX, participa en la diferenciación y migración neuronal, con la liberación del ácido ganmaminobutírico y glutamato que son importantes en estas funciones. Por lo tanto, las mutaciones en el gen STBP1 producen anormalidades en el tronco cerebral, muerte celular difusa. Los espasmos tónicos, típicos del síndrome de Ohtahara se consideran que ocurren como manifestación de afectación del tronco cerebral. Con el transcurso del tiempo estas anormalidades también originan las hipsarritmias, típicas del síndrome de West9.
En cuanto al origen de las descargas, se considera que se produce por una falta de regulación cortico-subcortical, debido a lesiones corticales. En estudios anátomopatológicos e inmunohistoquímicos se han hallado en pacientes fallecidos con el síndrome de Ohtahara, lesiones severas a nivel de putámen, tálamo, hipocampo y tegmento del tallo cerebral9.
El pronóstico de los pacientes portadores del síndrome de Ohtahara es pobre, ya que las convulsiones intratables conducen a un severo retraso psicomotor y cognitivo, y sobreviene generalmente la muerte en la infancia temprana16.
Es importante efectuar la distinción con la encefalopatía mioclónica temprana, que también se puede presentar dentro de los primeros tres meses de vida, a veces en el periodo neonatal, pocas horas después de nacer. Sin embargo, la presentación clínica de las convulsiones es por lo general como mioclonías focales en extremidades, cara, o un área de estos sitios como los dedos o los ojos. Los temblores se presentan en zonas aisladas del cuerpo en forma asincrónica, iniciándose en un sitio y migrando a otros. En el 80% de casos se presentan como convulsiones focales, por ejemplo, desviación de la mirada o posturas tónicas. También pueden desarrollar espasmos tónicos focales o de grupos musculares; a veces las manifestaciones son sutiles signos autonómicos, como rubor facial o apneas.
Con respecto a la manifestación en el electroencefalograma, también presentan el patrón de descargas-supresión, pero éstas aparecen con más frecuencia durante el sueño, y no son continuas. Es necesario destacar que el patrón típico puede no estar presente inicialmente, requiriéndose del seguimiento para descubrir su aparición. El patrón de ondas-supresión puede transformarse en hipsarritmia a los 3 a 5 meses en forma temporal, para posteriormente volver al patrón anterior9,16.
Como en el síndrome de Ohtahara, la encefalopatía mioclónica temprana puede ser manifestación de alteraciones estructurales del sistema nervioso, trastornos metabólicos o genéticos. Debemos señalar, sin embargo, que las alteraciones estructurales no son vistas inicialmente en éste síndrome, sino más bien en el curso de la enfermedad como una atrofia cortical. Este hallazgo sugiere la existencia de un desorden metabólico o degenerativo subyacente que posteriormente conduce a la alteración estructural9,16.
Entre los trastornos metabólicos relacionados con esta entidad se hallan la hiperglicemia no cetósica, acidemia diglicérica, aciduria propiónica, deficiencia del cofactor molibdeno, deficiencia de piridoxina, acidemia metilmalónica, deficiencia de sulfito oxidasa, síndrome de Menkes, y síndrome de Zellweger9.
En cuanto a los hallazgos patológicos en este síndrome, generalmente son trastornos difusos que afectan la substancia blanca y el tronco cerebral, lo cual conduce a trastornos de la diferenciación y a una hiperexitabilidad de la corteza. Las alteraciones patológicas halladas incluyen desmielinización, cambios espongiformes en la substancia blanca, alteración en la laminación de las capas corticales profundas de la corteza, cuerpos concéntricos perivasculares, y proliferación de astrocitos. También es frecuente la visualización de cordones neuronales en la substancia blanca, sugiriendo un trastorno de la migración neuronal y apoptosis como mecanismo subyacente9.
Los estudios de neuroimágenes muestran hipoperfusión e hipometabolismo en los ganglios basales, tálamo; y durante las descargas aumento de la perfusión en estos sitios, además del tronco cerebral y la corteza frontoparietal; lo cual orienta a la existencia de descargas de origen en estructuras subcorticales9.
Por otra parte, se describen algunos casos familiares en esta enfermedad, originando un cuestionamiento sobre la existencia de algún componente genético responsable de la misma9,16.
Debemos señalar que el diagnóstico de estas entidades se efectúa con el cuadro clínico y el hallazgo en el electroencefalograma. Los estudios de imágenes ponen en evidencia la presencia de alteraciones estructurales si es que existen. Los potenciales evocados del tronco encefálico en general son anormales en ambas afecciones.
Las drogas antiepilépticas, como el fenobarbital, fenitoína, valproato, zonisamida, y benzodiacepinas, tienen limitada utilidad en estas patologías. La hormona adrenocorticotropa resultó útil en el manejo de pacientes con síndrome de Ohtahara que evolucionan a síndrome de West. Es de destacar que ninguna de estas medicaciones resulta efectiva en el tratamiento de pacientes con encefalopatía mioclónica temprana; e incluso se ha descripto empeoramiento de algunos casos tratados con vigabatrina.
La dieta cetogénica ha resultado útil en algunos casos de síndrome de Ohtahara. La corrección del desorden metabólico subyacente puede conducir a mejores resultados en los casos asociados a esa patogénesis, como en la deficiencia de piridoxina, y biotinidasa. La encefalopatía mioclónica asociada a hiperglicemia no cetósica, puede responder al uso de benzoato de sodio, ketamina, dextrometorfano; algunas veces en combinación con triptófano, estricnina e imipramina. Sin embargo, se debe aclarar que estos tratamientos si bien pueden mejorar el curso neonatal, no modifican el resultado a largo plazo.
Los casos asociados a anormalidades estructurales operables, pueden ser tratados con resecciones neuroquirúgicas focales, como en los casos de hemimegalencefalia con hemiesferectomía17,18.
Por otra parte, la secuencia de Pierre Robin se caracteriza por la combinación de micrognatia y retrognatia, glosoptosis y distress respiratorio, con o sin defecto de paladar. La incidencia estimada es de 1 en 8500 a 14000 nacimientos; se asocia con alta morbilidad secundaria al compromiso de la vía aérea, dificultad para la alimentación y problemas del lenguaje11,12. Si bien la presentación clínica es característica, la patogenia de esta entidad no está completamente entendida. En alrededor de la mitad de los casos se presenta en forma aislada. Los casos aislados se considera que se deben a constricción intraútero del feto, por fuerzas extrínsecas (oligoamnios, anatomía anormal del útero), que impide el crecimiento normal de la mandíbula. La micrognatia en el temprano desarrollo fetal contribuye a que la lengua permanezca entre los esbozos que formarán el paladar, impidiendo de esa manera la fusión de los mismos. Sin embargo, este mecanismo ha sido refutado por algunos autores, ya que se ha visto que puede asociarse a delesiones cromosómicas tales como 2q24.1-33.3, 4q32-qter, 11q21-23.1, y 17q21-24.311. Recientemente se han encontrado también mutaciones en SOX9 y 17q24 asociados al síndrome (19,24.
Debemos resaltar que frecuentemente el síndrome de Pierre Robin se asocia con hallazgos clínicos que pueden constituir parte de un síndrome reconocido. Entre los síndromes más comúnmente asociados se mencionan el síndrome de Stickler, el síndrome velo-cardio-facial, que presentan mutaciones en 22q11.2, el síndrome alcohol fetal, la trisomía 18, y el síndrome Aicardi19-24.
Otro síndrome descripto es el acro-cardio-facial, en el cual la característica distintiva es la deformidad de las manos y pies, que puede ser unilateral o bilateral; caracterizada por subluxación de la articulación metacarpofalángica, con flexión en la articulación interfalángica proximal, puede existir ausencia de dedos, dedos cutáneos, anormalidad de los metacarpianos, sindactilia en dedos de los pies, hallus valgus. Sumado a esto, otra anormalidad presente en dos tercios de los pacientes es el defecto cardiaco, que puede consistir en defectos septales, defectos cono-truncales, o lesiones obstructivas del lado izquierdo. En cuanto al aspecto facial, pueden presentar frente amplia, ojos prominentes, puente nasal aplanado, orejas de implantación baja, fisura de labio y paladar. A nivel genital, los de sexo masculino pueden presentar micropene, criptorquidia, hipospadia. A nivel neurológico, se cita la presencia de retardo mental, hipotonía, hipertonía, convulsiones, atrofia cortical, quistes neuroepiteliales cerebrales, hipoplasia del cuerpo calloso. Como puede verse el espectro de defectos que pueden presentarse es amplio, existe variabilidad interindividual; se considera que es de herencia autosómica, ya que se presenta en casos de consanguinidad entre los padres y se ha visto recurrencia en hermanos en algunas familias. Se desconoce aún cuál es el gen comprometido. Se trata de una enfermedad muy rara, con incidencia aproximada de 1 en 100.000 recién nacidos25. Cabe resaltar que en el caso que se presenta, se desconoce si existe parentesco entre los padres del paciente.
El caso que estamos analizando tiene algunos datos que recuerdan al síndrome acro-cardio-facial, por el hallazgo de micropene, defecto de labio y paladar, convulsiones, retardo mental, y alteraciones en la estructura cerebral. Sin embargo, carece de defectos cardiacos y de las extremidades que son característicos del mencionado síndrome.
Por otra parte, se citan publicaciones de casos de asociación de síndrome de Pierre Robin con presencia de neurofibromas a nivel cerebral, en médula, y shawnomas de nervios craneales, como el octavo par, y convulsiones26. Teniendo como base mutaciones en genes. En este aspecto, debemos señalar que la madre del paciente presentaba hipoacusia, lo cual dificultaba la comunicación con el cuerpo médico encargado del cuidado de su hijo, desconocemos cuál es la etiología de dicha alteración auditiva.
También existe la publicación de una entidad presentada en dos hermanas de la presencia de síndrome de Pierre Robin con una imagen de megacisterna magna, secundaria a hipoplasia cerebelar, con importante retraso en el desarrollo psicomotor, además de deformidades en los dedos, camptodactilia, rigidéz en articulaciones de muñeca, codo, tobillo y rodilla, ojos inclinados hacia abajo, coloboma, nariz pequeña antevertida. Aunque no se identificó el gen comprometido, se piensa que se trata de una entidad con transmisión autosómica recesiva27.
CONCLUSIÓN
Aunque no hemos podido ubicar al paciente dentro de un determinado síndrome, consideramos importante describir el caso por lo poco frecuente, y por presentar un fenotipo que sugiere la existencia de algún transtorno cromosómico subyacente. Penosamente debemos admitir que esta pesquisa no pudo llevarse a cabo en el caso presentado, por no disponer en el Hospital y por la dificultad para el traslado del paciente a los centros que sí cuentan con medios para realizar estudios cromosómicos.
Finalmente, con el apoyo de publicaciones existentes podemos concluir que en los pacientes con convulsiones difíciles de controlar, se impone efectuar además de todo el grupo de investigaciones acostumbradas (imágenes, electroencefalograma, etc.), un estudio cromosómico y genético, en algunos casos también considerar la necesidad de realizar estudios metabólicos, ya que pueden existir alteraciones cromosómicas o metabólicas que expliquen la presencia de las mismas, y su reconocimiento permitirá orientar a los padres sobre el riesgo de recurrencia en futuros hijos. Lo cual es sumamente importante, sobre todo considerando el mal pronóstico que presentan estas afecciones.

