











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCION
Un nuevo patógeno hace su aparición en la escena mundial generando caos, pérdidas de vidas humanas y económicas, desconcertando a los profesionales de la salud. Nueva información surge todos los días a un ritmo que resulta imposible de analizar. Mucha de esta información se basa en anécdotas, observaciones y estadísticas. La presión social y mediática por encontrar una solución es importante. Se hace reiterada referencia a que en el COVID-19 se da una patogenia atípica, llamativa, poco habitual, pero pocos artículos o revisiones tratan de entender su fisiopatología, piedra fundamental para plantear cualquier tratamiento más o menos racional. En pos de entender al COVID-19 y desenredar los mecanismos patogénicos involucrados he realizado una revisión en la literatura centrado básicamente en cuatro aspectos: el virus y la respuesta inmune que desencadena, el sistema angiotensina y su regulación, la fisiopatología de la hipoxemia y los mecanismos de compensación, y el síndrome de distrés respiratorio del adulto (ARDS). Luego presento una posible clasificación fisiopatológica y recomendaciones terapéuticas en cada caso.
El Patógeno y la Respuesta Inmune
Un nuevo virus apareció en China a fines del año 2019, perteneciente al grupo de los coronavirus, virus caracterizados por poseer una “corona” de receptores de superficie que participan en la interacción con proteínas en las células del huésped. Este nuevo coronavirus se asocia a neumonía y fallo respiratorio y es muy similar al coronavirus que en el año 2003 afecto otra zona de China, el SARS-CoV, infectando a unas 8000 personas y matando a casi el 10% de estas 1. Por estas similitudes se nombra al nuevo coronavirus como SARS-CoV2, y la enfermedad que produce como COVID-19 (COronaVIrus Disease 2019).
El contagio ocurre por interacción con secreciones mucosas de una persona portadora o enferma con el virus, sea a través de interacción directa entre personas o por vía de objetos contaminados. El virus aparentemente sobrevive un tiempo prudente en el medio ambiente, siendo la infectividad mayor que la del SARS-CoV1, debido a diferencias en las proteínas de la “corona”, lo que junto a la gran proporción de portadores poco o nada sintomáticos, ha facilitado el desarrollo de la actual pandemia.
La corona del SARS-CoV2 está formada por las glucoproteínas S (spikes) que interactúan con el principal receptor del virus en células humanas, la enzima convertidora de angiotensina tipo 2 (ACE2), también receptor del SARS-CoV1. La glucoproteína S del SARS-CoV2 resulto ser antigénicamente diferente a la del SARS-CoV1 y poseen una afinidad 4 veces superior por la enzima ACE2 2, aunque otros investigadores han publicado datos diferentes 3. Para potenciar el ingreso del virus a la célula el complejo ACE2-proteína S debe ser modificado por la proteasa de membrana TMPRSS2, paso que puede ser bloqueado por anticuerpos o fármacos 4. Debido a este factor, las células más sensibles a la infección son las que expresan ambas proteínas, ACE2 y TMPRSS2. En el proceso de ingreso del virus a la célula huésped se produce una reducción marcada de la actividad ACE2 en superficie celular, tanto por pérdida al ingresar con el virus como por aumento de la escisión de la proteína y la liberación de la forma soluble sACE2 5. Posteriormente se identificó a una proteína de la familia de las inmunoglobulinas, el CD147, EMMPRIN o basigina, presente en eritrocitos y puerta para el ingreso del agente de la malaria, como otro receptor del SARS-CoV2. Algunos autores han conjeturado que el virus ingresa a los eritrocitos y altera la hemoglobina 6, lo que no ha sido confirmado y parece difícil dado que los eritrocitos maduros no realizan endocitosis debido a su citoesqueleto especializado. Los linfocitos T expresan tanto ACE2 como CD147 por lo que el ingreso del virus por estas vías podría ser la causa de la linfopenia reconocida en la COVID-19, sumada a otras casusas posibles como apoptosis en el contexto de una tormenta de citoquinas o como consecuencia de la elevación de glucocorticoides 7. La linfopenia parece ser un indicador de la carga viral y está asociada a peor evolución 8. En el SARS se constata un hallazgo similar, también asociado a peor evolución 9.
No hay datos que hagan referencia a un daño celular directo mediado por el virus. Dicho esto, surge la interrogante de cual es o son los mecanismos por los cuales se desarrolla insuficiencia respiratoria y ARDS como consecuencia de la infección por SARS-CoV2. En general los virus poseen tres mecanismos patogénicos: lesión celular directa, lesión a través de la respuesta inmune evocada o modificación de una vía fisiológica. Como se trata de un patógeno reciente aún se siguen acumulando datos sobre su ciclo vital, pero se pueden hacen algunas inferencias al observar la patogenia del SARS-CoV1, agente del SARS, cuadro muy similar al actual COVID-19 10. Aplicando este razonamiento todo parece indicar que los mecanismos patogénicos del SARS-CoV2 involucran inicialmente la alteración de vías fisiológicas centradas en torno a la enzima ACE2, a lo que se agrega posteriormente la respuesta inflamatoria.
Las primeras medidas contra el ingreso y multiplicación de los virus son orquestadas por integrantes del sistema inmune innato, incluidas células inmunes como células dendríticas, linfocitos NK y macrófagos, y no inmunes como células epiteliales y fibroblastos. La respuesta innata se basa en la detección de material biológico extraño (PAMPs) o de material derivado del daño celular (DAMPs). Estas sustancias son detectadas por los llamados receptores de reconocimiento de patrones (PRR), presentes tanto en la superficie celular como en endosomas, citosol y superficie mitocondrial. Los tres tipos más comunes de PRR son los TLRs (Toll-like receptors), los RLRs (RIG-I-like receptors) y los NLRs (NOD-like receptors). La activación de los PRR produce una cascada de señales intercelulares que termina por activar factores de transcripción involucrados en la defensa y supervivencia celular, como NF-κB e IRFs, factores que promueve la inflamación como un fenómeno de retroalimentación positiva. Conviene recordar que todo fenómeno de retroalimentación positiva es controlado o detenido por la introducción de “frenos” en alguna de las fases y que dejados sin control solo acaban cuando se agotan sus vías. Los incendios forestales son una buena analogía, donde pasado un punto ya no se puede extinguir activamente y solo queda esperar a que no haya más nada que quemar.
La activación de PRRs en células inmunes y no inmunes y la activación de NF-κB promueve la producción y secreción de las citoquinas IL-6 y TNFα, la activación de caspasas la secreción de IL-1β y la activación de los IRFs la producción de interferones (IFNs) 11,12. Estos últimos interfieren con el ciclo reproductivo del virus mientras que las citoquinas evocan cambios locales y sistémicos en pos de la eliminación de células infectadas, la reparación de tejido dañado y la producción de sustancias y células con actividad antimicrobiana, generando además cambios hemodinámicos y metabólicos que soporten dichas acciones. Estas citoquinas actúan por receptores de membrana que al ser activados generan nuevamente la activación del NF-κB y más secreción de citoquinas, llegando eventualmente a generar la llamada “tormenta de citoquinas”, que, como su nombre lo infiere, produce más daño que beneficio. Los IFNs actúan de forma autócrina y parácrina a través de receptores de membrana, produciendo de nuevo una respuesta proinflamatoria en la célula blanco, activando NF-κB y la producción de citoquinas, incluidos nuevamente los IFNs 13. El IFN-I promueve la síntesis celular de varias proteínas con actividad antiviral como viperina (CIG5), que interfiere con la producción de proteínas virales en el RE y la brotación de los virus formados, MXA, que forma oligómeros alrededor de particular virales, y IFITMs, que interfieren con la fusión del virus, ribonucleasas y otras 14.

Gráfico 1. El SARS-CoV2 activa diferentes receptores de reconocimiento de patrones que desencadenan la liberación de citoquinas inflamatorias. Los bucles de retroalimentación positiva que se generan pueden ser modulados por macrófagos M2, linfocitos T reguladores y la secreción de IL-10
El ciclo de retroalimentación positiva prosigue hasta que el patógeno y las células infectadas son erradicas o se produce el agotamiento del sistema inmune, sus células y mediadores o se genera daño suficiente como para comprometer la vida del organismo infectado. En este sentido la naturaleza es pragmática, destruye las células infectadas para eliminar el patógeno y salvar al individuo, pero destruirá al individuo si no puede erradicar el patógeno para salvar a la especie. Si la batalla evoluciona a favor del huésped el ciclo proinflamatorio es frenado por la activación de células antiinflamatorias como macrófagos M2 y LT reguladores (TREG), con secreción de IL-10 y estimulación de la reparación tisular (VEGF, TGFβ).
La evolución ha dotado a las células y organismos de medidas contra los virus, siendo muy efectivas en la mayoría de los casos, a diferencia de las intervenciones farmacológicas antivirales. Algunos virus sin embargo han evolucionado y generado contramedidas que potencian su supervivencia, replicación y contagio, sin matar rápidamente al organismo que los porta, lo que sería un despropósito desde el punto de vista del virus. El SARS-CoV1 comparte el casi el 80% de la información genética con el SARC-CoV2 15 y presenta un comportamiento clínico similar, aunque parece ser más contagioso, pero con una mortalidad menor, asociada también a “tormenta de citoquinas” y ARDS 15. El SARS-CoV1 expresa, como el SARS-CoV2, una glucoproteína S a través de la que interactúa con la enzima ACE2, así como proteína M, varias proteínas no estructurales (nrp) y proteínas involucradas en replicación viral. Estas otras proteínas han evolucionado para evitar la activación de los RLRs, la generación de IFN-I o la cascada que la activación de su receptor genera 14, así como la transcripción de mRNA de la célula huésped, monopolizando el sistema de síntesis de proteínas celular. En el pulmón la principal célula afectada por el SARS-CoV1 y por el SARS-CoV2 es el neumocito tipo II, célula de regeneración y encargada de la síntesis de surfactante.
La inmunidad adquirida posee también un papel en la infección por estos coronavirus. Se ha reportado que luego de la infección por SARS-CoV2 se puede encontrar IgM antes de la semana e IgG antes del décimo día, así como linfocitos T y NK contra el virus 15, probablemente dirigidos contra la glucoproteína S de la superficie viral. El papel de los anticuerpos contra la glucoproteína S está por determinarse, demostrándose, al menos en modelos animales, que su presencia podría ser más perjudicial que benéfica 5, coincidiendo la seroconversión con el peor momento de la enfermedad, y colocando en tela de juicio la eficacia de una vacuna. Este daño mediado por anticuerpos podría ser secundario a hiperactivación macrofágica en los alveolos o a daño celular mediado por anticuerpos. Además, se ha planteado la posibilidad que un título inadecuado de anticuerpos, por debajo del necesario para detener la diseminación viral, podría ser perjudicial al facilitar la entrada del virus en células que no poseen su receptor primario, pero si receptor para la porción Fc de las inmunoglobulinas, algo similar a lo que ocurre en el dengue 5, proceso denominado amplificación viral dependiente de anticuerpos (ADE, antibody-dependent enhancement) y que ha sido confirmado en otros coronavirus 16.
En la COVD-19 se observa linfopenia en más del 80% de los pacientes hospitalizados y más del 90% de los que requieren ingreso a terapia intensiva 17, y este hallazgo está ligado a mal pronóstico 7, sea por reducción de la respuesta inmune adquirida defensiva o por pérdida de la regulación de la inflamación. El SARS-CoV2 puede infectar linfocitos a través del receptor ACE2 y del receptor CD147 18, pero aparentemente no se replica en los linfocitos ni media su apoptosis, por lo que la causa de la linfopenia probablemente este asociado a la tormenta de citoquinas y a la elevación de los glucocorticoides.
En resumen, el SARS-CoV2 infecta células a través de la interacción de su glucoproteína S con proteínas de la superficie celular, en particular ACE2, pero también CD147, requiriendo además a la proteasa TMPRSS2. En este proceso el virus produce reducción de los ACE2 de superficie y aumento de los sACE2. El virus posee proteínas que reducen la respuesta por las células infectadas, pero de todas formas genera una respuesta inflamatoria creciente, que puede llegar al rango de tormenta de citoquinas. La generación de una respuesta humoral es máxima del décimo día, momento en el cual se puede dar un empeoramiento del cuadro debido probablemente a una potenciación mediada por anticuerpos. La linfopenia es un marcador de gravedad y mal pronóstico, quizás mediada por el ingreso del virus a los linfocitos T o, más probablemente, a la propia tormenta de citoquinas.
Sistema Angiotensina
El sistema renina-angiotensina-aldosterona (RAAS) es un sistema de regulación homeostático diseñado para oponerse a la pérdida de volumen efectivo circulante y a la caída de la presión arterial que esto genera. La desregulación del RAAS se asocia a hipertensión, daño oxidativo, disfunción endotelial, remodelado cardiovascular e inflamación. Inicialmente compuesto por las enzimas renina y ACE, la angiotensina II, sus precursores y receptores, la aldosterona y su receptor, en el año 2000 se le suma un nuevo integrante al identificarse la enzima ACE2 19, pasando a llamarse ACE1 a la primera identificada. Posteriormente se identificó la formación tisular de angiotensinas, con implicaciones tanto locales como sistémicas 20, así como nuevos receptores.
En la versión clásica del RAAS el sistema percibe el descenso del volumen circulante a través del aparato yuxtaglomerular de la nefrona, por cambios en el flujo sanguíneo renal y en la composición del líquido tubular, y/o por estímulo simpático como parte del baro-reflejo arterial. La caída de la presión arterial detectado por estos mecanismos promueve la secreción plasmática de la proteasa renina desde las células yuxtaglomerulares que habitan la pared de la arteriola aferente. La renina convierte el angiotensinógeno, una proteína circulante de 452 aminoácidos secretada por hepatocitos y adipocitos, en el decapéptido angiotensina I o Ang1-10, el que posteriormente puede ser sustrato de la acción de la dicarboxipeptidasa ACE1, enzima constitutiva del endotelio pulmonar, produciéndose angiotensina II o Ang1-8, principal efector del sistema. La AngII actúa a través de receptores de membrana del tipo GPCR, el AT1R (AT1AR y AT1BR) que activa la kinasa PKC, promueve la liberación de calcio desde el retículo endoplasmático y la activación de las enzimas NOX generadoras de especies reactivas del oxígeno (ROS), y el receptor AT2R que estimula la actividad de enzimas NOS generadoras de óxido nítrico (NO). El estímulo de los AT1R se asocia a vasoconstricción, remodelado vascular y cardíaco, daño oxidativo, insulino-resistencia, hipertensión, disfunción endotelial e inflamación, mientras que el estímulo AT2R posee acciones opuestas 21.
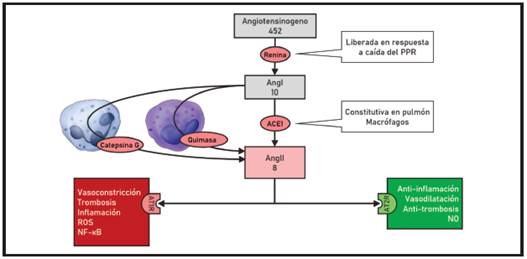
Gráfico 2. La AngII puede ser producida a partir de la secuencia renina-ACE1 o de forma independiente a partir de proteasas derivadas desde células inflamatorias. Sus acciones se realizan a través de dos receptores de membrana, con efectos opuestos
La AngII puede ser producida de forma independiente a la ACE1 por enzimas como quimasa (mastocitos, endotelio y otros) y catepsina G (neutrófilos) 22. La hipoxemia potencia la producción de AngII a través de la acumulación de lactato y la activación de la quimasa, a la par que la AngII refuerza la generación de ROS y activación del factor HIF-1α estimuladas por la hipoxemia 23. La AngII es sustrato de la carboxipeptidasa ACE2 que produce el péptido activo Ang1-7. Las aminopeptidasas producen AngIII y AngIV a partir de la AngII. Las modificaciones sobre la AngII por estas otras enzimas tienen un efecto global inhibitorio al reducir su concentración y generar angiotensinas con efecto fisiológico opuesto.
Más allá del efecto homeostático sobre medio interno y presión arterial que tiene la AngII, y de los mentados efectos deletéreos que genera su elevación crónica sobre la función cardiovascular, cada vez más se hace mención del papel de la AngII circulante y parácrina sobre la respuesta inmune e inflamación. La AngII ejerce acciones pro-inflamatorias actuando sobre el receptor AT1R, promoviendo la producción de ROS y la activación del factor NF-κB 24, la expresión endotelial de moléculas de adhesión, la secreción de quimocinas para monocitos como la MCP-1, la presentación de antígenos, y la activación y migración de linfocitos T(21). Tanto los mediadores inflamatorios como la AngII alterar la fisiología endotelial promoviendo el cambio fenotípico hacia la actividad pro-inflamatoria, pro-coagulante 25 y de aumento de la permeabilidad vascular, en detrimento de sus funciones de barrera, anti-coagulante y vasodilatadora. Las citoquinas inflamatorias a su vez potencian la formación de AngII al elevar la producción hepática de angiotensinógeno y la actividad ACE1, y reduciendo la actividad ACE2 26. Los neutrófilos además poseen enzimas como la catepsina G capaz de generar localmente AngII a partir de AngI 27. La AngII ha sido implicada en varias patologías como diabetes mellitus y esclerosis múltiple, pero de particular interés es la asociación con el ARDS en donde su elevación, junto con la reducción de la Ang1-7, se ha asociado a desarrollo de distrés y mayor mortalidad 28, así como su bloqueo o reducción se ha implicado en mejoría al menos en algunos estudios y modelos animales 29.
La ACE o ACE1 es una dicarboxipeptidasa de membrana celular, dependiente de zinc, con 2 dominios catalíticos, expresada en endotelio pulmonar, epitelio intestinal y de túbulos renales, macrófagos y neutrófilos 30. En macrófagos y neutrófilos la mayor expresión de ACE1 se relaciona con una mayor respuesta inflamatoria y defensiva, que es independiente a la producción de AngII y está asociada a una mayor liberación de citoquinas y producción de ROS 30. Los niveles de AngII son particularmente elevados en las enfermedades granulomatosas como sarcoidosis y tuberculosis. La ACE1 es responsable de producción AngII a partir de AngI y del catabolismo de las bradicininas, sustancias vasodilatadoras que estimulan la actividad de la eNOS. La AC1E también inactiva la Ang1-7, principal producto de la ACE2 y antagonista funcional de la AngII 31. El gen que codifica la ACE1 posee polimorfismo y tres variantes posibles, siendo el genotipo D/D el que posee mayor tasa de transcripción y el asociado a mayor riesgo de enfermedades metabólicas y cardiovasculares.
La ACE2 es una monocarboxipeptidasa de membrana celular, dependiente zinc, pero con un solo dominio catalítico. Se expresa principalmente en epitelio intestinal pero también en epitelio respiratorio, en especial en neumocito tipo II 32, endotelio, testículo, placenta, mucosa orofaringea 33 y epitelio nasal 34. De particular interés resulta la expresión de ACE2 en núcleos del tronco como el NTS y la asociación entre su reducción y la amortiguación de la respuesta baro-receptora en modelos animales 35. La ACE2 puede sufrir sección de su ancla de membrana por la ADAM17 36, generando una forma soluble o sACE2. La ACE2 posee alta afinidad por la AngII a la que hidroliza generando el péptido activo Ang1-7, sin actividad contra las bradicininas, pero si contra otros péptidos como la ghrelina 36. La importancia de su actividad es doble, por un lado, reduce los niveles circulantes de AngII y por otro genera otra molécula, la Ang1-7, que a través de un receptor específico genera acciones contrarias a la de la AngII. La ACE2 produce además a partir de la AngI la Ang1-9, un agonista del receptor AT2R y con acciones también contrarias a las de la AngII. Es así como se genera una suerte de yin-yang, por un lado, la ACE1/AngII, y por otro la ACE2/Ang1-7 y Ang1-9, con acciones sobre endotelio, vasos y respuesta inflamatoria opuestas.
La expresión de ACE2 en adipocitos y epitelio renal es promovida por la proteína FGF21 37 liberada por hepatocitos en respuesta a cambios en la dieta, por adipocitos (adipocitoquina) en respuesta a temperaturas ambientes bajas y por rabdomiocitos en respuesta a ayuno y ejercicio (miocitokina). La reducción de la FGF21 o su resistencia y elevación, como la observada en obesidad, se asocia a enfermedades metabólicas, inflamación e hipertensión arterial. La potenciación de la actividad ACE2 se encuentra entre las medidas potencialmente más eficaces para el tratamiento de la hipertensión arterial pulmonar 38.
Otra enzima importante en la generación de angiontesinas opuestas a la AngII es la neprisilina (NEP), una endopeptidasa de membrana ubicua conocida por su papel en el catabolismo de los péptidos natriuréticos. La NEP puede generar Ang1-7 a partir de Ang1-9 y también a partir de AngI como lo hace la ACE2 39.
La Ang1-7 actúa sobre un receptor denominado Mas (por Massey, apellido del donante humano del tejido donde fue identificado) 40. El MasR es un GPCR expresado en endotelio, corazón, riñón y cerebro, que activa la cascada PI3K/Akt y la actividad de las enzimas NOS. Por otra parte, la Ang1-7 se une a AT1R antagonizando los efectos de la AngII al activar la vía de la β-arrestina 41 que concluye con la internalización del receptor y la activación de cascadas diferentes a las activadas por AngII, como son de nuevo la vía PI3K/Akt y la síntesis de NO. En varios modelos animales de sepsis, daño pulmonar agudo (ALI) y de distrés pulmonar (ARDS) se confirma el desbalance ACE1/AngII vs ACE2/ Ang1-7 a favor del primero, con reducción del proceso inflamatorio al utilizar inhibidores ACE1 (IECAs) o bloqueantes AT1R (ARAs) 22. En pacientes hipertensos se ha demostrado una menor eliminación urinaria de Ang1-7 en relación con sus cifras tensionales, indicando una menor actividad renal de ACE2 38. Como se comentó previamente la infección por SARS-CoV1 y SARS-CoV2 reduce la expresión de ACE2 y de por sí mueve la balanza hacia el lado ACE1/AngII, promoviendo a inflamación, vasoconstricción pulmonar y edema, ARDS y lesión endotelial. En modelos animales de ARDS se ha confirmado la relación entre descenso de la actividad ACE2, elevación de la actividad AngII, lesión pulmonar e hipoxia 19. La pérdida de la actividad ACE2 podría además promover las lesiones cardíacas, renales y cerebrales, en particular en los centros cardioreguladores y respiratorios del tronco 31. La utilización de bloquentes ACE1/AngII o infusión de Ang1-7 podría ser benéfica al inicio de la patología, pero perjudicial si ya se ha desarrollado una falla hemodinámica.
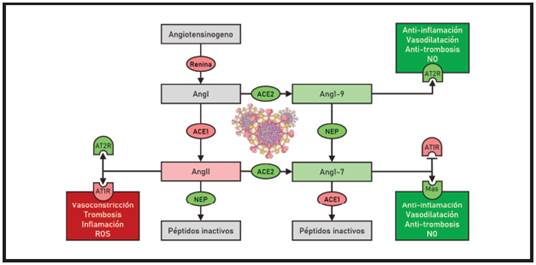
Gráfico 3. La enzima ACE2 participa en la formación de angiotensinas con acciones opuestas a las de la AngII que median sus acciones a través de los receptores MasR y AT2R. El SARS-CoV2 reduce la formación de estos antagonistas
La disfunción endotelial hace referencia a la pérdida de la integridad de la barrera que presenta el endotelio entre compartimientos vascular e intersticial, sumado a la reducción de su capacidad vasodilatadora, antitrombótica y antiinflamatoria. Muchas de estas funciones son mediadas por la producción continua de NO por parte de la eNOS, enzima cuya actividad es estimulada por el flujo sanguíneo y la cascada PI3K/Akt, parte de las acciones mediadas por insulina y Ang1-7. En este punto cabe recordar que uno de los principales factores de riesgo para el desarrollo de complicaciones debidas a la infección por SARS-CoV2 es la diabetes mellitus, quizás debido a que se suman la resistencia a la insulina con la reducción de la Ang1-7, lo que profundiza la disfunción endotelial. Los ROS generados por la mitocondria en situaciones de hipoxia o generados por el estímulo de enzimas NOX por AT1R, posee el efecto contrario al NO, reduciendo su disponibilidad y promoviendo el cambio del fenotipo endotelial hacia el aumento de la permeabilidad (HIF-1α y VEGF), la adherencia de leucocitos y la actividad procoagulante (NF-κB).
El COVID-19 que requiere hospitalización e ingreso a terapia intensiva es más frecuente en mayores de 60 años, en varones (60%), hipertensos (60%), obesos (40%) y diabéticos (30%) pero no, llamativamente, en fumadores, asmáticos o enfermos pulmonares crónicos 42, teniendo casi el 90% de los pacientes dos o más comorbilidades. Ya se ha implicado a la hiperfunción de ACE1/AngII en la patogenia de la hipertensión arterial, la diabetes mellitus y la obesidad 20, por lo que es válido pensar que las personas con estas dolencias poseen un desequilibrio a favor de ACE1/AngII en contra de ACE2/Ang1-7, colocándolas en especial riesgo de desarrollar complicaciones fruto de la profundización de dicho desequilibrio por la infección con SARS-CoV2. Recordar además que el exceso de AngII y/o reducción de Ang1-7 ha sido implicada en varios estudios en la patogenia de ALI y ARDS 29 así como en la mentada disfunción endotelial. Más allá de cruda estadística podemos intuir que los pacientes con mayor riesgo de complicaciones parecen ser los que poseen los clásicos riesgos cardiovasculares, recordando la repetida frase de especialistas en cardiología que dice que el endotelio en el blanco de dichos factores, lo cual sería también cierto en estos pacientes, ya que el endotelio es blanco, directo o mediado por citoquinas, hipoxemia o anticuerpos, del SARS-CoV2.
Desde que la identificación de la enzima ACE2 como principal puerta de ingreso del SARS-CoV2 a la célula ha habido preocupación por el consumo de bloqueantes ACE1/AT1R debido a informes experimentales de que estos fármacos aumentan la expresión celular de la enzima ACE2, sin existir confirmación de este hallazgo en humanos. Por otra parte, estudios experimentales han demostrado beneficio con estos fármacos en prevención y tratamiento del ARDS 43. Debido a la falta de información de calidad al respecto la comunidad científica no se decide y por el momento no recomienda la inclusión de estos fármacos como parte del tratamiento del COVID-19, aunque tampoco recomienda su suspensión en pacientes que ya lo consumen. Paulatinamente sin embargo la balanza comienza a inclinarse a favor de la utilización de bloqueantes ACE1/AT1R 44 en pacientes con COVD19 mientras no existan contraindicaciones obvias. Un estudio reciente no encontró mayor riesgo de infección con SARS-CoV2 o peor evolución de la enfermedad en pacientes tratados con estos fármacos 45.
Resumiendo, la actividad de la ACE1, los niveles de AngII y la activación del receptor AT1R se asocia a hipertensión arterial sistémicas y pulmonar, vasoconstricción, daño oxidativo mediado por ROS, aumento de la permeabilidad vascular, así como de los fenómenos inflamatorios y trombóticos, habiendo sido involucrados en patologías como ARDS, hipertensión pulmonar, obesidad y síndrome metabólico. Las diferencias entre los niveles interpersonales de AngII puede ser fruto de diferencias genéticas en el gen de la enzima ACE1 o en los niveles del sistema de contra-regulación formado por las enzimas ACE2 y NEP, los péptidos Ang1-7 y Ang1-9, y los receptores MasR y AT2R. Este sistema opuesto no solo metaboliza y reduce los niveles de AngII, sino que a través de sus receptores ejerce acciones opuestas como vasodilatación, reducción de la respuesta inflamatoria y estabilización endotelial, mediados en parte por aumento de la producción de NO. En el COVID-19, como en el SARS, existe un desequilibrio a favor de la AngII debido a la reducción de la actividad de la ACE2, probablemente más acusado en personas portadoras de algunas comorbilidades como obesidad, hipertensión y diabetes, con participación en la patogenia del ARDS y en complicaciones vasculares como los accidentes cerebrovasculares 46.
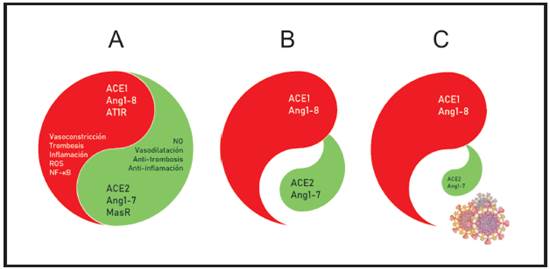
Gráfico 4. Equilibrio ACE1/ACE2. En el esquema A existe un equilibrio entre las acciones de la ACE1, la AngII y su receptor, por un lado, y la ACE2, la Ang1-7 y el receptor MasR por el otro. En B se observa un desequilibrio a favor de lado que promueve inflamación y trombosis como se observa en pacientes que presentan obesidad, hipertensión, diabetes mellitus u otras causas de disfunción endotelial. Como se muestra en C la infección por SARS-CoV2 profundiza dicha disparidad potenciando el proceso inflamatorio y la aparición de trombosis
La Hipoxemia
La saturación normal de la hemoglobina con oxígeno (SatO2) en sangre arterial, es mayor a 95% respirando aire ambiente (FiO2 0.21) a nivel del mar (1 atm o 760 mmHg). Con una ventilación pulmonar habitual (unas 12 respiraciones por minuto, moviendo 500 ml de aire en cada ciclo) y un espacio muerto habitual (ventilación no utilizada para intercambio) se entrega una ventilación alveolar de más de 4 litros por minuto, lograndose una pO2 alveolar (pAO2) y arterial (paO2) de unos 100 mmHg. El CO2 difunde 20 veces mejor que el O2 a través de la membrana alveolo-capilar, asociándose la hipercapnia a hipoventilación alveolar, sea por una hipoventilación pulmonar deficiente (obesos, por ejemplo) o por un aumento de la ventilación desperdiciada (aumento del espacio muerto). Con una concentración de hemoglobina normal en sangre y una SatO2 normal se transportan en sangre arterial unos 20 ml de O2 cada 100 ml de sangre. Es importante recordar que la pO2 (presión parcial de oxígeno) hace referencia al O2 disuelto en los líquidos corporales y no al unido a la hemoglobina. Se define como hipoxemia a una pO2 menor a 80 mmHg o una SatO2 menor a 95% en sangre arterial. Con una pO2 menor a 60 mmHg, o una SatO2 menor a 90%, la hipoxemia se considera moderada. Las causas de hipoxemia incluyen reducción de la pO2 ambiente (grandes alturas o incendios), reducción de la ventilación (acompañada de aumento de la pCO2), alteración de la relación ventilación/perfusión o V/Q (la causa más frecuente), alteración de la difusión (reducción de la superficie de intercambio o aumento del grosor de la membrana de difusión) o por cortocircuito de derecha a izquierda (exceso de alveolos no ventilados, síndrome hepatopulmonar, cardiopatías cianotizantes).
Las manifestaciones de la hipoxemia dependen de su grado y del tiempo de instauración, habiendo variaciones interindividuales. Los síntomas pueden deberse a la respuesta automática compensadora o a la disfunción de órganos que requieren una pO2 estable para su función normal como cerebro o corazón. La hipoxemia aguda leve (SatO2 90-94%) suele generar taquipnea y disnea (hambre de aire), así como taquicardia reactiva. En la hipoxemia aguda moderada se incrementa la disnea y taquicardia y se agrega síntomas de encefalopatía incipiente (mareos, alteraciones visuales y auditivas), astenia, mialgias y cefalea (vasodilatación cerebral). En la forma severa el paciente progresa hacia el coma y al fallo circulatorio por disfunción cardíaca y vasodilatación sistémica.
La respuesta homeostática ante la hipoxemia se inicia en los cuerpos carotideos, órganos minúsculos especializados en el monitoreo de la pO2 arterial 47. Los cuerpos carotideos se ubican en la bifurcación de la arteria carótida primitiva, midiendo un par de milímetros y pesando unos pocos gramos, son considerados los órganos con mayor flujo sanguíneo en relación con su peso, flujo fundamental no solo para “sentir” la pO2 arterial sino porque poseen una elevada tasa metabólica. Reciben nervios motores simpáticos que regulan el flujo sanguíneo del órgano, y por ende su sensibilidad, y terminaciones aferentes que llevan la información por el nervio del seno carotideo (Hering), al nervio glosofaríngeo, y al núcleo del tracto solitario (NTS) en el tronco cerebral, información que luego regula centros ventilatorios y autonómicos. La sensibilidad de las células glómicas tipo I, principales células de los cuerpos carotídeos, a los cambios de pO2 puede ser modulada a su vez por los cambios de pH y pCO2 en sangre. Es importante mencionar que los cuerpos carotídeos son reactivos a los cambios de pO2, pero no a los cambios en el total de O2 transportado 48, siendo insensibles a la anemia, la hemodilución o la intoxicación por CO, salvo que estas alteraciones produzcan hipoxia celular y acidosis. Tampoco son sensibles a la extracción aumentada de O2 en periferia como ocurre en la piel por frio o hipotensión, lo que podría evidenciarse clínicamente por una SatO2 baja en la oximetría de pulso a pesar una SatO2 y pO2 normal en sangre arterial. El mecanismo por el cual la hipoxemia produce estímulo de las células glómicas y aumento de la frecuencia de potenciales de acción transmitidos es un tema aún en discusión. Se admite que la hipoxemia reduce la permeabilidad al potasio al cerrar, directa o indirectamente sus canales 49, generando despolarización, apertura de canales de calcio sensibles a voltaje y exocitosis de vesículas con neurotransmisores como ACh y ATP. Se ha demostrado la expresión de ACE1 y ACE2 en el cuerpo carotideo, así como la expresión de AT1R y MasR, considerándose a la AngII un potenciador de su actividad 50. Dada la expresión local de este sistema, particularmente ACE2 51, es válido pensar que la fisiología y regulación de este sensor fundamental se afecta durante la infección por SARS-CoV2.
La resección de los cuerpos carotideos fue una técnica introducida en la década del 60 como parte del tratamiento de pacientes con asma o enfermedad pulmonar obstructiva crónica sin respuesta a tratamiento médico con el fin de reducir la disnea y mejorar la calidad de vida de estos pacientes 52, técnica no exenta de oposición y dudas. En la década de los 80 surgen reportes que comunican la mejoría de la disnea en pacientes sometidos a esta cirugía, a pesar de una reducción de la pO2 y leve elevación de la pCO2, asociado a la reducción del volumen minuto a expensas de una menor frecuencia respiratoria 53. Más recientemente se ha reevaluado la utilidad de esta intervención en reducir el tono simpático en pacientes hipertensos con apnea del sueño, así como reducir la disnea en pacientes con insuficiencia cardíaca 54. Aunque no se aclara el mecanismo en la mejoría de la disnea con la resección de los cuerpos carotideos se puede inferir de estos hallazgos que la disnea no se presenta como respuesta primaria a la hipoxemia, sino que surge por estímulo de los centros respiratorios a partir de la percepción de dicha hipoxemia por los cuerpos carotideos o a partir de información sensitiva proveniente de los músculos respiratorios en respuesta al aumento de la ventilación.
La percepción de disnea, que posee componentes emocionales individuales, no se relaciona directamente con los gases en sangre arterial sino más bien con el aumento de las eferencias motoras hacia músculos respiratorios, en especial hacia músculos respiratorios accesorios, así como aferencias sensitivas que informan sobre el aumento en el uso de dichos músculos, su fatiga o acidosis 55. Por ello puede existir disnea sin hipoxemia, como ocurre durante el ejercicio físico o una crisis de pánico, o hipoxemia sin sensación de disnea si por alguna razón se evita el aumento de la respuesta ventilatoria, como ocurre experimentalmente en sujetos bajo los efectos de bloqueantes neuromusculares o, como se comentó, por lesiones de los cuerpos carotídeos. Algunos estudios demuestran que la activación de quimiorreceptores centrales y zonas límbicas por aumentos de la pCO2 puede generar la sensación de disnea, independiente a la respuesta de los músculos respiratorios. La activación de los mismos centros límbicos puede ser la causa de la sensación de disnea en las crisis de pánico. Globalmente se considera que la elevación de la hipercapnia es un estímulo más potente de la sensación de disnea que la hipoxemia 56, por lo que las situaciones en donde se desarrolla hipoxemia sin hipercapnia, como ser en los cortocircuitos de derecha a izquierda no acompañados de cambios en la complacencia o volúmenes pulmonares, no producen disnea manifiesta. En el alpinismo sucede algo similar, con caída progresiva de la pO2 alveolar y arterial a mayor altura, pero sin aumento de la pCO2 arterial. Otro estímulo disneico, aunque aparentemente secundario, parecen ser las fibras C del intersticio pulmonar que responden a irritación o estiramiento y transmiten a través del nervio vago, quizás con algún papel en la disnea del edema intersticial o las neumonitis 57. La reducción de la complacencia pulmonar, por reducción del surfactante, edema o fibrosis intersticial, supone un sobreesfuerzo de los músculos inspiradores y, como tal, se asocia a la sensación de disnea.
La respuesta sistémica a la hipoxemia detectada por los cuerpos carotideos se gesta en núcleos autonómicos del tronco cerebral. El llamado quimio-reflejo promueve el aumento de la ventilación minuto, así como aumento del estímulo vagal sobre corazón y del estímulo simpático sobre los vasos de la musculatura, con el fin de reducir el consumo de O2. La hiperventilación reactiva estimula los mecano-receptores pulmonares que reducen la descarga cardíaca vagal y la descarga vasoconstrictora muscular, por lo que habitualmente se observa taquicardia sin grandes cambios en la presión arterial. Cuando la hiperventilación reactiva no ocurre, como en la apnea del sueño o en la apnea del “reflejo de sumersión”, se constata bradicardia y elevación de la presión arterial por aumento del tono simpático vascular. La respuesta ventilatoria a la hipoxia posee base genética, con diferencias interpersonales, además de variaciones diarias individuales.
En el pulmón también se da una respuesta vascular ante la hipoxia, una vasoconstricción que pretende, en el mejor de los casos, derivar la sangre a unidades respiratorias más ventiladas, mejorando la relación V/Q y el intercambio gaseoso. Es importante aclarar que solamente los vasos sanguíneos pulmonares reaccionan ante la hipoxia con vasoconstricción, con el agravamiento paradójico de la hipoxia en las unidades respiratorias que la desarrollan. Si este proceso ocurre solo en pocas unidades alveolares la presión pulmonar se mantiene debido a que la resistencia global de la circulación pulmonar es baja. Regionalmente existen variaciones de la V/Q incluso en personas sanas, con alta V/Q en vértices y más baja en las bases del pulmón en una persona de pie. En patologías pulmonares agudas localizadas, como una neumonía lobar, la respuesta vasoconstrictora mantiene la relación V/Q cercana al ideal de 0,8 58 y evita la hipoxemia, minimizando el cortocircuito de derecha a izquierda al evitar que la sangre circule por zonas no ventiladas. Las arteriolas pulmonares reaccionan a la pO2 local, suma de pO2 alveolar (pAO2) y la pO2 de la sangre venosa mixta (pvO2) 59, predominando una presión sobre la otra según halla más ventilación o más perfusión. El mecanismo molecular por el cual la hipoxia produce contracción de los miocitos lisos vasculares en arteriolas pulmonares no ha sido dilucidado por completo, pero se asemeja a la respuesta de las células glómicas del cuerpo carotideo, la hipoxia produce el cierre de uno o varios tipos de canales de K+ con la consiguiente despolarización e ingreso de calcio, pudiendo estar involucrados la mitocondria y la producción de ROS. Esta respuesta es potenciada por acidosis, hipertermia, ferropenia, angiotensina II, sistema simpático y endotelina-1, y reducida por alcalosis, NO y prostaciclinas 59. La vasoconstricción por hipoxia posee un papel patogénico en el desarrollo de edema pulmonar de altitud (HAPE), ALI y ARDS.
Me parece conveniente hacer un comentario aparte sobre la hipoxia hipobárica o “hipoxia de montaña” y sus consecuencias debido a la polémica que ha suscitado la comparación entre el “pulmón de montaña” o HAPE y la neumonía de la COVID-19. A medida que se asciende en altura la presión atmosférica y la pO2 del aire inspirado descienden, generando una caída de la pAO2 y paO2 progresivamente mayor. Por ejemplo, en Bogotá, ubicada a más de 2600 m, la pO2 ambiente es cercana a 120 mmHg y la pAO2 de unos 60 mmHg, mientras que, en La Paz, a unos 3000 m, la pO2 ambiente se reduce a 110 mmHg y la pAO2 a unos 55 mmHg. Las complicaciones de la hipoxia de altura son mayores cuanto más rápido se asciende, son sobretodo cerebrales y pulmonares, se desarrollan en los primeros 4 días y se relacionan con la elevación de la actividad simpática, la presión arterial pulmonar y el flujo sanguíneo cerebral 60. Las complicaciones cerebrales incluyen al “mal de montaña” y al “edema cerebral de altura”, patologías en donde la vasodilatación cerebral reactiva y la hipoxemia producen edema cerebral, hipertensión endocraneana, coma y eventualmente la muerte 61. La complicación pulmonar es el HAPE que se manifiesta inicialmente como tos y astenia, para producir luego disnea y esputo rosado 61. La radiografía de tórax en la forma avanzada evidencia infiltrado alveolar parcheado, asimétrico, periférico y bilateral, pero inicialmente puede ser normal. Los principales factores de riesgo incluyen el ascenso rápido, es decir un rápido desarrollo de la hipoxemia, haber desarrollado la condición previamente, y ser obeso, en particular si existen trastornos ventilatorios 62. Las personas que no desarrollan una respuesta ventilatoria acorde y no compensen la hipoxemia presentan un mayor riesgo de desarrollar tanto edema pulmonar como edema cerebral de altura 62. La respuesta ventilatoria a la hipoxia es mayor en jóvenes y se reduce después de los 50 años. El principal fenómeno patogénico en el edema pulmonar de altura es el desarrollo de una poderosa respuesta vasoconstrictora pulmonar que, al ser parcheada, produce estrés funcional en los capilares con mayor flujo sanguíneo, potenciando la salida de líquido y edema intersticial, con lesión final de la barrera alveolo-capilar, escape de plasma y hemorragia alveolar. La hipoxia, y su persistencia por una anormal respuesta ventilatoria, genera inicialmente aumento del tono simpático, vasoconstricción en arteriolas pulmonares y aumento de la presión arterial pulmonar, lo que, sumado al menor aclaramiento de líquido alveolar por una menor actividad de la bomba de sodio y potasio en el epitelio alveolar, facilita la salida de líquido hacia intersticio y el alveolo, empeorando la hipoxemia. Eventualmente la hipoxia persistente inicia una respuesta inflamatoria, con acumulación de neutrófilos y liberación de citoquinas. En este punto la inflamación y la hipoxia se potencian mutuamente, pudiendo desarrollarse un ARDS.
El tratamiento inicial del HAPE se basa en reducir la vasoconstricción por hipoxia, al administrar O2 y utilizar vasodilatadores pulmonares como los bloqueantes de la PDE3, tadalafil por ejemplo, y los bloqueantes ACE1/AT1R. Otras medidas incluyen el uso de dexamentasona y β2AR agonistas, los que parecen potenciar el aclaramiento alveolar. Estas medidas, probadas útiles en el tratamiento del HAPE, no han resultado beneficiosas para el tratamiento del ARDS, básicamente porque ninguna de estas medidas reduce la inflamación y su retroalimentación positiva. De hecho, el uso de vasodilatadores pulmonares en el ARDS puede empeorar el fenómeno de cortocircuito y agravar la hipoxemia 63, por lo que no están indicados.
Desde el inicio de la pandemia se ha comparado al pulmón de COVID-19 con el HAPE y sin lugar a duda poseen puntos en común, con la diferencia que el ingrediente inflamatorio aparece antes en la COVID-19 como reacción al patógeno y no como reacción exclusiva ante la hipoxia como en el HAPE. Declarar tajantemente que no tienen absolutamente nada que ver 64 implica no haber analizado la fisiopatología de ambos procesos. Se ha hecho referencia a pacientes hipoxémicos sin síntomas por lo que una posibilidad es que el SARS-CoV2 inicialmente altere la difusión de O2 sin generar una respuesta inflamatoria manifiesta, debido a disfunción de pneumocitos y comprometimiento del aclaramiento alveolar y de la producción de surfactante, reduciendo la ventilación, así como al producir cambios vasculares al potenciar los efectos de la AngII, potenciando la respuesta vasoconstrictora y alterando la perfusión. En este escenario se producirá una reducción de la paO2 sin cambios en la paCO2, cambios que serán más pronunciados en personas con una deficiente respuesta ventilatoria a la hipoxia, como personas mayores de 50 años y obesos. Este escenario hipoxémico no inflamatorio inicial es muy similar al HAPE, donde los fenómenos vasculares predominan, y quizás sea la razón de la similitud entre los hallazgos radiológicos entre HAPE y neumonía del COVID-19, procesos que eventualmente evocan una respuesta inflamatoria suficiente llegando a generar ARDS. Por ello no se puede comparar ARDS del COVID-19 con HAPE, sino sería más racional comparar ARDS del COVID-19 con ARDS por HAPE. Es probable que la infección viral acelere la aparición de una respuesta inflamatoria y que el desarrollo de ARDS en el COVID-19 sea más precoz y frecuente que secundaria a HAPE.
A nivel celular la hipoxemia activa varios mecanismos de adaptación, dirigidos a ahorrar energía y reducir el consumo de O2, fomentar la glucolisis como fuente de energía y reducir el estrés oxidativo. Actores centrales en estos procesos son los factores inducidos por hipoxia o HIFs, que promueven la angiogénesis, el estímulo de la respuesta ventilatoria, la eritropoyesis y los cambios metabólicos necesarios para ahorrar O2. Los HIFs funcionan como heterodímeros de una unidad HIFα (1 o 2) y una HIFβ que se unen a genes con promotores con secuencia HRE (Hypoxia Response Element). En normoxia la subunidad HIFα es continuamente hidroxilada por enzimas PHD (Prolyl Hydroxylase Domain), proceso sensible a los niveles de O2 que requiere además vitamina C, hierro ferroso y α-keroglutarato. Los factores prolil-hidroxilados son reconocidos y ubicuitinizados por un complejo que incluye a la proteína pVHL (Von Hippel-Lindau), con la consiguiente proteólisis en proteosoma. Otra hidroxilasa, la FIH (Factor Inhibiting HIF1), también sensible a los niveles de O2, impide la actividad estimulante de la transcripción del dímero HIF 65. En hipoxia las PHD dejan de estar activas por lo que HIFα puede formar dímero con HIFβ, ingresar al núcleo celular y activar la transcripción de genes con promotores HRE. Las PHD pueden inactivarse de forma independiente a la hipoxia, por ejemplo, por modificación oxidativa vía ROS o por fallo de la fosforilación oxidativa y acumulación de metabolitos como succinato y fumarato 65. La hipoxia genera otras respuestas celulares independientes de la activación de HIFs, como son la activación de la kinasa PERK en respuesta a estrés oxidativo en retículo endoplasmático y la inhibición del complejo mTORC1, ambos con la misma consecuencia, reducción de la traducción de proteínas y del gasto energético celular. En la mitocondria la hipoxia genera acumulación de ROS y liberación de Ca2+, que puede activar las kinasas CaMKK y AMPK, sensor energético que reduce aún más los procesos que consumen ATP. Los HIFs estimulan la transcripción GLUT1 y de enzimas glicolíticas, como LDH y PDK, que desvían el piruvato fuera de la mitocondria, amortiguando la producción de ROS 65 y potenciando la liberación de lactato. Un dato a favor de la participación central de la hipoxia y los HIFs en la COVID-19 es la presencia de angiogénesis pulmonar en pacientes fallecidos con este diagnóstico, a diferencia de los pacientes fallecidos por influenza, sumado a la presencia de microangiopatía y trombosis que coloca al endotelio vascular en el centro del escenario patogénico 66.
Hipoxia e inflamación se estimulan mutuamente en muchos niveles 67,68. Durante la inflamación la activación del factor de transcripción NF-κB, que coordina la transcripción de citoquinas, quimocinas y moléculas de adhesión, se activa a través de la cascada iniciada desde PRRs o de receptores de citoquinas, promoviendo la transcripción de HIFα al tiempo que estimula la producción de ROS y reduce el hierro disponible, inhibiendo así a las PHDs. El aumento de transcripción de los HIFs posibilita su acción mantenida en el tiempo. Los HIFs promueven cambios vasculares proinflamatorios al estimular la producción de VEGF, conocido por desestabilizar la barrera endotelial, al fomentar cambios metabólicos activadores en macrófagos y neutrófilos 67 y reducir su apoptosis 68. La hipoxia a su vez promueve la activación del factor NF-κB por mecanismos no del todo resueltos que podrían involucrar a PHD/FIH inhibiendo IKK (kinasa del inhibidor IκB), así como la liberación de calcio desde la mitocondria con activación de CaMK, TAK1 y del complejo IKK 69. La activación del factor NF-κB durante la hipoxia evita la apoptosis y fomenta la angiogénesis, reforzando las acciones de los HIFs. Como en general los tejidos con respuesta inflamatoria intensa sufren de hipoxia, es comprensible que estos factores se activen para reforzar la respuesta inflamatoria, aumentar la resistencia celular y mejorar el flujo sanguíneo, echando leña al fuego por así decirlo. Una prueba de que la hipoxemia como único agresor produce inflamación se encuentra en alpinistas que desarrollan mal de altura y edema pulmonar, con elevación de interleucina 6 y PCR 68. Otros estudios han demostrado la asociación de hipoxia y daño pulmonar agudo inflamatorio 70,71. Todo indica que le respuesta inflamatoria evocada por la hipoxia es más severa en el pulmón que en otros órganos, en parte porque los alveolos están habituados a una tensión de O2 muy superior a lo de otros tejidos, y en parte por la respuesta vasoconstrictora ante la hipoxia, particular de la circulación pulmonar, que puede agravar la hipoxia local y generar lesión.
Desde el inicio de la pandemia de COVID-19 se ha comunicado la existencia de pacientes que se presentaban a la sala de emergencia con hipoxemia por gasometría de pulso, pero sin referir disnea, bautizando este fenómeno como “hipoxemia silente o hipoxemia feliz”. En las series de características clínicas de los pacientes internados que se han publicado hasta la fecha 17,42, no hay datos de gases en sangre, por lo que el concepto gira en torno a la oximetría de pulso. Más allá de lo “curioso” del fenómeno ha sido asociado con peor evolución de los pacientes.
La causa no está definida, pero podría ser por una menor respuesta a la hipoxemia por disfunción de los cuerpos carotideos o de los centros nerviosos del tronco ya que ambos expresan ACE2. Sea cual fuera la razón la falta de disnea se puede entender como reducción de la quimio-reflejo y de la respuesta ventilatoria. La falta de compensación de la hipoxemia evocaría una respuesta vascular pulmonar más intensa, potenciada por la inflamación y la reducción de la expresión de ACE2 generada por la infección viral. Una respuesta vascular pulmonar intensa genera, como se comprueba en el HAPE, un edema pulmonar parcheado por aumento del filtrado capilar, y eventualmente lesión alveolar.
Resumiendo, se define como hipoxemia a una pO2 menor a 80 mmHg o una SatO2 menor a 95% en sangre arterial, siendo su causa principal las alteraciones V/Q. Los síntomas de la hipoxemia dependen de su intensidad y de la velocidad de su desarrollo, así como de factores individuales y son derivados inicialmente de la respuesta compensadora pero luego son consecuencia de la hipoxia celular. La hipoxemia se percibe en los cuerpos carotideos, desencadenando una serie de reflejos con centros en el tronco cerebral que incluyen el aumento del tono simpático vascular y el aumento de la ventilación pulmonar. Los cuerpos carotideos no perciben la SatO2 sino la paO2. Estos órganos poseen un RAS local, que incluye la ACE2, pudiendo verse funcionalmente afectado durante la infección por SARS-CoV2, habiéndose demostrado que su extirpación o lesión genera reducción de la sensación de disnea y de la respuesta compensadora ventilatoria. Hipoxia e inflamación son capaces de crear un bucle de estímulo mutuo, centrado en los HIFs, los ROS y el NF-κB, siendo el pulmón el órgano más sensible a la hipoxia y donde esta produce una respuesta inflamatoria mayor. En la circulación pulmonar la hipoxia genera vasoconstricción con el fin de mejorar la relación V/Q. El lado oscuro de este mecanismo es que, por un lado, agrava la hipoxia de las zonas poco ventiladas y por otro aumenta el estrés hemodinámico de las zonas ventiladas. La respuesta vasoconstrictora es estimulada por AngII y el sistema simpático y si es intensa o persistente puede generar edema pulmonar y lesión de la barrera alveolo-capilar, iniciando una respuesta inflamatoria. Una mayor capacidad vasoconstrictora pulmonar junto con una reducción de la respuesta ventilatoria puede coexistir en pacientes mayores de 50 años, obesos y/o hipertensos, personas que están en mayor riesgo de desarrollar edema pulmonar en situaciones de hipoxemia. En la infección por SARS-CoV2 hay disfunción de neumocitos y cambios vasculares pulmonares por reducción de la ACE2, alteración de la V/Q y desarrollo inicial de hipoxemia que, de persistir, evoca una respuesta inflamatoria. Tanto en HAPE como en COVID-19 de persistir la hipoxia se produce una respuesta inflamatoria, con potenciación mutua entre ambas, generando finalmente un ARDS. La fisiopatología detrás de la “hipoxemia feliz” no está clara, pero puede deberse a una reducción de la respuesta ventilatoria por edad, comorbilidad o disfunción de los cuerpos carotídeos o centros del tronco por infección con SARS-CoV2, a vasoconstricción periférica por reducción de la actividad de la ACE2 y mayor extracción de O2 o una combinación de estos factores. Más allá de la causa, su existencia puede ser un signo de mala evolución si se asocia a una respuesta ventilatoria reducida.

Gráfico 5. La disnea surge como percepción del mayor esfuerzo respiratorio, es decir, es un síntoma de la respuesta compensatoria ante la hipoxemia. La ausencia de disnea puede deberse a falta de percepción de la hipoxemia por lesión en los cuerpos carotídeos o de los núcleos del tronco
Síndrome de Distrés Respiratorio
Este síndrome es el resultado de una respuesta inflamatoria autosustentable en los pulmonares, iniciada por una variada lista de situaciones, con la sepsis como la más frecuente. Entre las causas del ARDS (por sus siglas en inglés) se encuentran infecciones pulmonares virales, como la generada por H1N1 o el SARS-CoV1. Una vez establecido es muy difícil diferenciar el origen del ARDS, por lo que la base terapéutica suele ser la misma, con el agregado de una terapia específica si se conoce la causa. Aún con los avances tecnológicos y médicos actuales el ARDS tiene un pobre pronóstico, con una mortalidad entorno al 40% 72. El ARDS se define desde el 2012 por la presencia de los criterios de Berlin 73, que hacen referencia al edema pulmonar no cardiogénico con desarrollo de cortocircuito de derecha-izquierda intrapulmonar evidenciado por una relación entre paO2/FiO2 menor a 300 mmHg.
El evento fundamental en el ARDS es la pérdida de integridad de la barrera alveolo-capilar, con escape de exudado hacia intersticio y alveolo, infiltración con neutrófilos y formación de trombos, causando hipoxemia por alteración V/Q y cortocircuito de derecha a izquierda. La anatomía patológica en la fase aguda revela membranas hialinas, descamación del epitelio, infiltración neutrofílica, hemorragia alveolar, microtrombos y atelectasias por pérdida de surfactante. Hallazgos similares se encontraron en autopsias de pacientes fallecidos por COVID-19 74, reafirmando la presunción de que estos pacientes fallecían al desarrollar ARDS. Este patrón se denomina “daño alveolar difuso” (DAD) y se presenta en el 50% de los pacientes con diagnóstico clínico de ARDS, asociándose a peor pronóstico. El otro 50% sele evidenciar formas con menos lesiones de la membrana alveolo-capilar, pero con infiltrado neutrofílico alveolar. Clínicamente se puede presumir la existencia de DAD en pacientes con ARDS que presentan una relación paO2/FiO2 menor de 100 y compromiso radiológico en los cuatro cuadrantes 75.
El pulmón es blanco frecuente de enfermedades inflamatorias, sea como actor principal o sufriendo daño colateral. La susceptibilidad radica en una enorme red vascular, una velocidad de flujo sanguíneo lento y una red de capilares que permiten la adhesión y diapédesis de neutrófilos, cuando en otras redes vasculares este fenómeno ocurre en las vénulas poscapilares 76, sin comprometer por ende la oxigenación del tejido. El capilar pulmonar, de unos 10 μm o menos, comprime al neutrófilo de casi 15 μm de diámetro, que no necesita del “rodamiento” para adherirse. Los neutrófilos son movilizados en respuesta a quimocinas generadas desde el epitelio respiratorio o desde los macrófagos alveolares. Sea cual fuera el estímulo para la movilización de neutrófilos estos se impactan en la microcirculación pulmonar, sufren frecuente NETosis (red formada por la expulsión del material genético), activando plaquetas y promoviendo la formación de trombos. Los neutrófilos activan la formación local de AngII, potenciando los fenómenos vasculares. La hipoxia en los sitios inflamados genera activación de HIFs, con acción proinflamatoria y profundización de la alteración vascular. Finalmente se crea un bucle de retroalimentación positiva con el desarrollo de daño tisular evidenciado como DAD en la anatomía patológica. Como se ha comentado este no siempre es el caso, habiendo pacientes con el diagnóstico clínico de ARDS sin el desarrollo de DAD, que poseen mejor pronóstico. En busca de responder la interrogante de porque algunos pacientes desarrollan ARDS y otros no, se encontró la asociación de este síndrome con el genotipo DD del gen de la ACE1, asociada a mayor expresión de la enzima 77. También se ha demostrado el beneficio de la actividad ACE2 en el pronóstico del ARDS tanto en modelos animales 78,79, así como recientemente en estudios preclínicos humanos 80. Otro dato a favor de la participación central patogénica del disbalance ACE1/ACE2 en el ARDS lo aporta el hallazgo de concentraciones elevadas de AngII en pacientes con ARDS por COVID-19 8. Otros autores ya han comentado esta relación 81.
En el ARDS, desde antes de la actual pandemia COVID-19, se ha identificado el aumento del riesgo trombótico y la utilidad de la anticoagulación 82, así como el aumento del dímero-D y la ferritina como factores de mal pronóstico 83. La elevación de la LDH, enzima citoplasmática ubicua necesaria para la formación de lactato desde piruvato y la regeneración de NAD+ para la glicolisis, es un marcador inespecífico de muerte celular, como la que ocurre en tejidos inflamados, isquémicos, cánceres o hemolisis, pero también se eleva en respuesta a la hipoxia y activación de los HIFs 65. La inflamación y la disfunción endotelial, junto con la formación de microtrombos y la hiperactividad macrofágica, son causales de hemolisis, liberación de LDH y de hemoglobina. La hemoglobina libre se fija a la haptoglobina y el complejo puede ser reconocido por el receptor CD163 en macrófagos, sufriendo endocitosis y metabolización, liberando ion ferroso y estimulando la producción de ferritina. La hemoglobina que escapa a este mecanismo de limpieza genera daño al atrapar NO y generar vasoconstricción, promover la activación de neutrófilos y al actuar como un DAMP, estimulando la actividad pro-inflamatoria y pro-trombótica del endotelio 84. La elevación de la ferritina en procesos como el ARDS se debe en parte a la activación de macrófagos mediada por los productos derivados de la hemólisis microangiopática 85. Para algunos autores la disfunción endotelial genera una “enfermedad vascular por microtrombosis”, con hallazgos similares a los de la púrpura trombótica trombocitopénica siendo el ARDS la manifestación pulmonar de esta patología sistémica 85. La ferritina reduce el estrés oxidativo de la célula al minimizar la presencia de ion ferroso libre y la posibilidad que se forme el nocivo radical hidroxilo (reacción de Fenton). Los macrófagos bajo influjo de citoquinas aumentan, por activación NF-κB, la producción de ferritina. Independientemente a la causa desencadenante, la hiperferritinemia se asocia a aumento de mortalidad y es reflejo de la activación macrofágica, liberación de citoquinas y hemolisis microvascular. La elevación del dímero-D supone trombosis y fibrinólisis intravascular, algo frecuente en el ARDS debido a la inflamación, la disfunción endotelial, el aumento de la síntesis de factores de la coagulación y la activación de plaquetas a través de la interacción con neutrófilos en la microcirculación pulmonar. La inmunotrombosis, la trombosis potenciada por citoquinas y células inflamatorias, es un proceso frecuente en el ARDS independiente de su causa 86, facilitado por el desequilibrio ACE1/ACE2 y al aumento de la formación de AngII, que no solo posee efectos pro-inflamatorios sino además pro-trombóticos 87. Esta trombofilia asociada al ARDS puede ser la responsable del aumento de fenómenos trombóticos venosos 82 y arteriales.
Armando el Rompecabezas
Es hora de juntar toda la información expuesta previamente, tratando de conformar un cuadro fisiopatológico coherente y que explique los diferentes hallazgos y formas patológicas del COVID-19. Para ello considero didáctico intentar separar en fases o estadios a la enfermedad.
En las formas leves y asintomáticas los afectados adquieren posiblemente un inóculo viral menor y no portan previamente un desequilibrio ACE1/ACE2, es decir, no son obesos, hipertensos, diabéticos o de edad avanzada. En este contexto la actividad ACE2 no desciende por debajo de un umbral crítico y las manifestaciones solo surgen de la respuesta inflamatoria local, siendo un proceso autolimitado y sin compromiso de la función pulmonar.
En las formas hipoxémicas compensadas un inóculo viral mayor podría generar disfunción del epitelio respiratorio, reduciendo la producción de surfactante y el aclaramiento de líquido alveolar, con aparición de alveolos poco ventilados. Esto sería balanceado por una respuesta vasoconstrictora pulmonar que desviaría la sangre hacia alveolos mejor ventilados. De persistir la hipoxemia por alteración V/Q los cuerpos carotideos estimularían una repuesta ventilatoria. Estos pacientes se presentarían taquipneicos, taquicárdicos pero con hipoxemia leve, y sin marcadores inflamatorios sistémicos. Estas formas pueden ser autolimitadas y no pasar a mayores, pero podrían agravarse si no se puede mantener la respuesta ventilatoria compensadora en el tiempo, como puede ocurrir en personas añosas o con patología pulmonar o cardíaca de base.
En las formas hipoxémicas descompensadas los pacientes que no desarrollan una respuesta ventilatoria acorde como consecuencia de una amortiguación de la respuesta a la hipoxemia por edad avanzada, disfunción preexistente de los cuerpos carotideos, o compromiso de la función de los cuerpos carotideos o del NTS mediada por el virus, desarrollan mayor hipoxemia con pocos o nulos síntomas inicialmente (hipoxemia feliz). La mayor hipoxemia y, lo que es peor, su permanencia, activa dos fenómenos. Por un lado, una mayor respuesta vasoconstrictora y por otro el inicio de una respuesta inflamatoria, que se agrega a la iniciada por la infección viral. La respuesta vasoconstrictora no solo se ve potenciada por la persistencia de la hipoxemia no compensada sino además por la reducción de la actividad ACE2 mediada por la infección. A su vez los individuos con una menor actividad ACE2, como ser adultos mayores, hipertensos, diabéticos y obesos, o mayor actividad ACE1 por portar el genotipo D/D, desarrollarán una respuesta vasoconstrictora mayor. En este punto el individuo desarrolla las lesiones pulmonares que han caracterizado al COVID-19 y que son muy similares al HAPE, con empeoramiento de la hipoxemia. La respuesta inflamatoria iniciada por el virus, eventualmente potenciada por el desarrollo de anticuerpos, es agravada por la hipoxemia y la reducción de la actividad ACE2. Dejada a libre evolución estos bucles de retroalimentación entre hipoxemia e inflamación se agravan.
La forma de afección multiorgánica compromete seriamente la vida. Llegado un punto el proceso inflamatorio se autosostiene, con producción elevada de citoquinas como IL-1β, IL-6 e interferones. Las citoquinas inflamatorias y la hipoxemia generan disfunción endotelial sistémica, pero probablemente de mayor intensidad en el pulmón, por las características particulares del órgano. En el pulmón la obstrucción microvascular con inmunotrombos, formados por neutrófilos y plaquetas, genera hemólisis y aumento del espacio muerto. En otros alveolos el aumento de la permeabilidad capilar, efecto de las citoquinas inflamatorias, del VEGF liberado por activación de los HIFs en respuesta a hipoxia y de la pérdida de la actividad anti-inflamatoria de la ACE2, junto con la descamación del epitelio respiratorio, la pérdida de aclaramiento alveolar y de surfactante, y una mayor presión hidrostática capilar fruto de la vasoconstricción pulmonar, generan la inundación alveolar con líquido rico en proteínas, neutrófilos y hematíes, mayor activación macrofágica y el desarrollo de cortocircuitos intrapulmonares, con agravamiento de la hipoxemia. Como la afección endotelial, secundaria a la tormenta de citoquinas, la reducción de ACE2 y la hipoxemia, es de tipo sistémico, pueden aparecer lesiones microvasculares o formación de trombos en otros órganos además del pulmón, como ser riñón y cerebro. El corazón puede ser blanco de esta disfunción microvacular, o verse afectado por la hipoxemia persistente, el aumento del tono simpático o los cambios hemodinámicos pulmonares. En este punto aparecen todos los hallazgos típicos de una respuesta inflamatoria con lesión tisular, como elevación de LDH, linfopenia, datos de eventos trombóticos, como el aumento del dímero-D, hemólisis y trombocitopenia, y de activación inmune como hiperferritinemia. Encaja muy bien en esta fase multiorgánica la conceptualización del ARDS como la manifestación orgánica de una enfermedad sistémica, la denominada enfermedad microtrombótica vascular, y no como causa de la misma, lo que explica la afectación frecuente de otros órganos en pacientes con diagnóstico clínico de ARDS (85). En línea con este concepto otros autores han asociado a las formas graves de COVID-19 con disfunción endotelial 88.
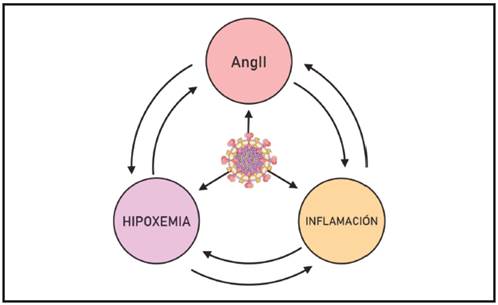
Gráfico 6. Existe una potenciación recíproca entre AngII, la hipoxemia y la inflamación, a través de varios mecanismos, en especial el estímulo de la producción de ROS y la activación del NF-κB. El SARS-CoV2 potencia los tres eventos
Recomendaciones
En cuanto al tratamiento, debería ser direccionado a la forma de la enfermedad. En la forma leve no se requiere tratamiento alguno más allá del reposo y del aislamiento. En la forma hipoxémica compensada se podría realizar tele-monitoreo del paciente con manejo ambulatorio, evaluando SatO2 y signos vitales en pacientes sin factores de riesgo. En pacientes con factores de riesgo la presencia de hipoxemia debe suponer monitorización y oxigenoterapia hospitalaria.
Las formas descompensadas requieren sin lugar a duda internación y optimización de la oxemia a través de terapias no invasivas y pronación. Queda por determinar si tendrían valor terapéutico los bloqueantes de la ACE1 o del receptor AT1R como indica el racionamiento fisiopatológico. De existir algún antiviral efectivo contra el SARS-CoV2 debería indicarse, así como una terapia específica contra otros agentes coinfectantes. En este respecto todo parece indicar que un antiviral de amplio espectro análogo de nucleótidos, el Remdesivir, será parte del armamento específico contra el SARS-CoV2. El uso de asistencia respiratoria mecánica en pacientes con COVID-19 está en revisión debido al hallazgo de una mortalidad mayor, cercana al 60%, que la esperada en pacientes con ARDS por otras razones. Una posibilidad es que en esta fase el fenómeno fundamental es el vascular y no el ventilatorio. Colocar a estos pacientes en ARM con una PEEP elevada comprometería aún más el flujo sanguíneo pulmonar, ya comprometido por la vasoconstricción excesiva en respuesta a hipoxia y pérdida de la actividad ACE2, al distender el alveolo y comprimir los vasos, así como al reducir el retorno venoso hacia el corazón derecho. Estudios recientes han demostrado que en el grupo de pacientes que requieren oxigenoterapia la dexametasona reduce la mortalidad, la necesidad de ARM y la estadía hospitalaria, quizás al reducir la respuesta inflamatoria, fomentar la producción de surfactante y mejorar el aclaramiento de líquido alveolar 89.
En la forma de COVID-19 asociada a ARDS y compromiso multisistémico la inflamación es la protagonista, y dado que es un proceso con retroalimentación positiva, dudo que los fármacos antivirales o los anticuerpos contra el virus posean beneficios importantes. Una prueba de lo poco efectivo que han sido las medidas terapéuticas en enfermedades inflamatorias como shock séptico y ARDS radica en su pobre pronóstico. En estas patologías la medicina moderna se enfoca en mantener vivo al paciente, con asistencia respiratoria mecánica, drogas vasopresoras, hemodiálisis y, eventualmente, mediante un sistema de oxigenación por membrana extracorpórea (ECMO), mientras la inflamación o se agota o se resuelve. Es evidente que medidas como la infusión de ACE2 o los inhibidores ACE1 o el tadanafil poco tienen que hacer en este escenario, como lo demuestran varios estudios, siendo sin embargo eficaces en prevenir el desarrollo de ARDS. La anticoagulación en esta fase parece razonable.
CONCLUSION
El centro de la patogenia del COVID-19 es la reducción de la actividad ACE2 frente a la ACE1, con mayor producción y efecto de la AngII en detrimento de los efectos de las angiotensinas antagonistas Ang1-9 y Ang1-7. Bajo esta premisa es obvio que los pacientes que evolucionen hacia las formas más graves de la enfermedad son aquellas que, previa a la infección, ya poseen una reducción de la expresión de ACE2 (adultos mayores, obesos, diabéticos) o sobre-expresan ACE1 al poseer genotipos particulares. La enzima ACE2 está involucrada en inmunidad, regulación de la función endotelial y regulación de la respuesta baro y quimio-receptora. En modelos animales y algunos estudios clínicos humanos se ha comprobado lo deletéreo de su reducción, así como los beneficios de su estímulo o infusión.
Para una mejor aproximación terapéutica de los pacientes con COVID-19 parece racional clasificarlos según el momento fisiopatológico. Cada uno de estos momentos o formas poseen opciones terapéuticas diferentes. En este sentido es primordial evitar el desarrollo de ARDS y de una respuesta inflamatoria sistémica, identificando los pacientes predispuestos y administrándoles terapias oportunas. El uso precoz de asistencia respiratoria mecánica en pacientes con la COVID-19 no ha demostrado buenos resultados, con una mortalidad entorno al 60% 90. Los protocolos de asistencia respiratoria mecánica para el tratamiento del ARDS, situación donde la pérdida de alveolos ventilados y la generación de cortocircuito es el fenómeno fundamental, no pueden ser aplicados en pacientes COVID-19 que inicialmente no desarrollan ARDS sino un edema pulmonar por vasoconstricción pulmonar y elevación de las resistencias pulmonares, similar a lo que ocurre en el HAPE.
Financiación propia.

