











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Tal vez se deba replantear el nombre de la fibrosis pulmonar familiar (FPF) ya que la misma describe a cuadros de fibrosis pulmonar idiopática (FPI) u otras neumonías intersticiales idiopáticas (neumonía intersticial no específica, neumonía intersticial descamativa, bronquiolitis respiratoria asociada a enfermedad pulmonar intersticial y neumonía organizante criptogénica) en dos o más miembros primarios de una familia biológica (padres, hijos o hermanos)(1). Dado que pueden darse distintas formas de compromiso parenquimatoso entre familiares es atractivo pensar en “neumonías intersticiales familiares” (NIF) como etiqueta más ajustada.
Un minucioso enfoque clínico de la FPI puede detectar 33% de casos relacionados en pariente próximo, cifra que pudiera aumentar a 53% con tamizaje mediante tomografía de alta resolución de tórax(2). Esto es tan estruendoso que, comparado a la historia de tabaquismo y la exposición ocupacional, la FPF constituye el factor de riesgo más importante para la presencia de FPI(3). Hoy se sabe que la FPF es una entidad que involucra mutaciones relacionadas con los telómeros o el surfactante pero es indistinguible clínica e histológicamente de la FPI(4).
La neurofibromatosis, la enfermedad de Niemann-Pick, la enfermedad de Gaucher, la disqueratoris congénita, la esclerosis tuberosa, la hipercalcemia hipocalciúrica familiar y otras entidades pueden cursar con NIF(5). En este reporte se describen las características clínicas de pacientes con el síndrome de Hermansky-Pudlak (SHP) y compromiso pulmonar vistos en un hospital de referencia del Ministerio de Salud de nuestro país, antes de la pandemia COVID-19.
CASOS CLINICOS
Caso 1. Varón de 45 años, con albinismo oculocutáneo, disminución de agudeza visual y hallazgos semiológicos casuales (crepitantes bilaterales) posterior a cirugía de fractura de antebrazo izquierdo. Disnea a grandes esfuerzos y referencias de hematomas espontáneos ante mínimos traumatismos de larga data. En la radiografía de tórax (no mostrada) se observan imágenes reticulonodulillares en base del hemitórax izquierdo que en la tomografía de tórax de alta resolución (TACAR) se observan como áreas de panalización periféricas y basales que empeoran en controles sucesivos. La broncoscopía no acotó datos de valor. Durante el seguimiento clínico, 9 meses desde el diagnóstico, la disnea empeora hasta mínimo esfuerzos. A la par de los síntomas clínicos, los parámetros radiológicos y funcionales se deterioran notoriamente (Figura 1), falleciendo aproximadamente a los dos años del diagnóstico por insuficiencia respiratoria.
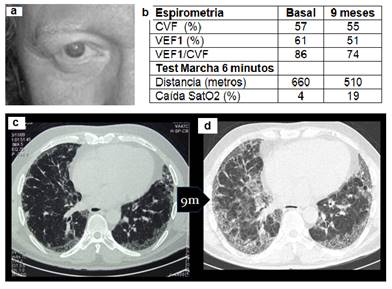
Figura 1. (a) Hipopigmentación de cejas, piel y ojos (albinismo oculo cutáneo); (b) Valores previstos de capacidad vital forzada (CVF) y del volumen espiratorio forzado en el primer segundo (VEF1) y resultados de la prueba de marcha de 6 minutos en consulta inicial y 9 meses después; (c) TACAR de tórax que muestra opacidades periféricas en segmentos periféricos junto a escasas áreas de panalización; (d) TACAR de tórax con mayor compromiso difuso, extensión de las áreas de vidrio esmerilado y presencia de panalización extendida con franca alteración de la arquitectura pulmonar.
Caso 2. Mujer 56 años, con albinismo oculocutáneo y nistagmo horizontal, hermana del paciente referido más arriba, refiere tos y disnea de 2 años de evolución. Antecedente de sangrado fácil y hematomas ante traumatismos mínimos desde la niñez. Antecedentes de cirugía con complicaciones hemorrágicas que requirieron transfusión (colelitiasis y cesárea). Crepitantes basales, SatO2 98% en reposo. La TAC de tórax se muestra en la Figura 2. Viaja al exterior con el fin de realizar estudios de trastornos plaquetarios y trazado genético, sin éxito. Desde el diagnóstico hasta los dos años de seguimiento tanto la clínica, las imágenes y las medidas de función pulmonar fueron deteriorándose progresivamente (Figura 2) hasta insuficiencia respiratoria refractaria.
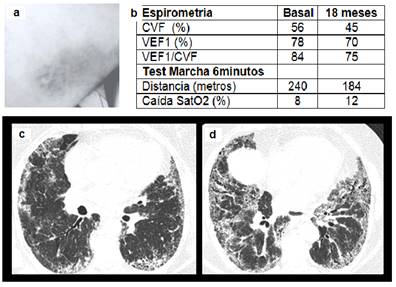
Figura 2. (a) Hematoma espontáneo en miembro superior; (b) Valores previstos de CVF y VEF1y resultados de la prueba de marcha de 6 minutos en consulta inicial y 18 meses después; (c) TACAR de tórax que muestra opacidades periféricas que en segmentos respetan espacios subpleurales con una bronquiectasia de tracción aislada y esbozos de panalización en región posterior; (d) TACAR de tórax con mayor compromiso difuso, áreas de vidrio esmerilado y extensión de las áreas de panalización y de las bronquiectasias de tracción con franca alteración de la arquitectura pulmonar.
DISCUSIÓN
El síndrome de Hermansky-Pudlak (SHP) es un trastorno autosómico recesivo poco frecuente causado por mutaciones que afectan los orgánulos relacionados con lisosomas. Clínicamente se caracteriza por la presencia de albinismo mucocutáneo y diátesis hemorrágica, aunque algunos pacientes pueden desarrollar colitis granulomatosa, grados variables de inmunodeficiencia y/o fibrosis pulmonar. Las pruebas genéticas tienen un rol fundamental en la confirmación de la entidad pero las limitaciones inherentes a su disponibilidad y costes, impulsan a considerar a las manifestaciones clínicas clásicas como indicadores relevantes del síndrome (6). En este reporte, el albinismo oculocutáneo es denominador común entre ambos hermanos así como la historia de sangrado fácil y hematomas espontáneos. Hay que considerar que la pérdida de pigmentación en el SHP es variable y depende del tipo genético determinante de modo que el color del cabello puede ir del blanco al marrón e incluso oscurecerse con la edad. El nistagmo horizontal y la mala agudeza visual se ven en la mayoría de los pacientes(7). Es relevante puntualizar que el cribado habitual para alteraciones de la crasis sanguínea puede ser normal(8).
Existen varios subtipos de SHP, causados por mutaciones en los genes humanos HPS1 a HPS10 que codifican proteínas formadoras de la biogénesis de los complejos de orgánulos lisosomales (BLOC), los cuales se pueden expresar como un desorden en los melanosomas (albinismo), cuerpos densos de plaquetas (discrasia sanguínea), cuerpos lamelares (fibrosis pulmonar) y gránulos líticos de linfocitos T citotóxicos (inmunodeficiencia)(9-10).
La fibrosis pulmonar ocurre siempre en pacientes adultos con mutación HPS1 (67 variantes patológicas), a veces en adultos con HPS4 (32 variantes) o en niños con HPS2 (29 variantes). No existen diferencias notorias en las manifestaciones respiratorias entre la FPI y la fibrosis pulmonar asociada a SHP (FPSHP), aunque algunos autores puntualizan que esta última acomete a personas en la cuarta década(11). Esto resalta la importancia de la historia clínica y, muchas veces, del interrogatorio reiterativo, ante la sospecha EPID en adultos con menos de 50 años. Creo particularmente que la nemotecnia M.O.P.C. (medicamentos, ocupaciones, primarias y colagenopatías) puede ser útil para la investigación de causas potenciales de EPID en atención primaria. Ante la presunción de FPF, es imperativo establecer los siguientes niveles de evidencia: antecedentes de fibrosis pulmonar idiopática o neumonitis intersticial no específica en parientes de primera línea (grado A), antecedentes de EPID no precisada (grado B), y antecedentes de enfermedad pulmonar no especificada (grado C)(12). La pesquisa del linaje no consigna historia de enfermedades respiratorias en consanguíneos de los pacientes aquí presentados aunque en primer grado consta un individuo con rasgos de hipopigmentacion al igual que otro familiar de cuarto grado en línea paterna. Solo en limitados centros de investigación del mundo existe la posibilidad de establecer los rasgos genéticos de los subtipos de SHP y los costos, conforme averiguado en época pre-pandémica, son mayoritariamente inaccesibles para nuestra población.
Una mayor precisión en el seguimiento clínico se consigue mediante las pruebas de función pulmonar y es de interés práctico realizar algunas consideraciones. Vemos en este trabajo que, ante la progresión de los síntomas, la CVF se modifica significativamente (>5%) solo en uno de los casos a lo largo del tiempo, mientras que en el test de marcha de 6 minutos las variables de interés (distancia recorrida prevista y caída de la saturación de oxigeno) parecen reflejar con mayor ajuste la progresión de los síntomas. Esto destaca la necesidad de incorporar rutinariamente las pruebas de ejercicio básicas a la rutina clínica de pacientes con enfermedades pulmonares difusas. Se recomienda monitoreo anual de la función pulmonar en individuos con mutaciones HPS1, HPS2 y HPS4, incluso desde la adolescencia (6). La capacidad de difusión del monóxido de carbono (DLCO) y la capacidad pulmonar total (CPT) suelen estar reducidas de modo variable en individuos con FPSHP leve (13) y se debe tener precaución en la interpretación pues hay que considerar la adecuación de valores a las ecuaciones de referencia utilizadas y la variabilidad intrínseca que puede presentar la medición de la DLCO. En pacientes con enfermedad pulmonar severa y con franca progresión se pueden constatar tasas de hasta 15% de disminución anual en los valores de CVF junto a hipoxemia reducida al esfuerzo con franco compromiso de los volúmenes pulmonares y de la capacidad de difusión(14).
Otro elemento muy útil para el monitoreo de la progresión de la FPSHP es la TACAR de tórax, cuyos hallazgos se correlacionan inversamente con la CVF(15). En estadios tempranos de la FPSHP, la tomografía realizada en el paciente en posición de decúbito prono discriminará áreas incipientes de enfermedad intersticial en regiones posterobasales. Las opacidades en vidrio esmerilado sobre un aspecto de neumonitis intersticial usual (panalización subpleural y basal, bronquiectasias de tracción y alteración en la arquitectura pulmonar) constituye un carácter discrepante del patrón clásico de la FPI y señala estadios más avanzados pudiéndose observar además reticulaciones bilaterales y engrosamiento septal en región subpleural y basal(16). El hallazgo de neumonitis intersticial en la TACAR de tórax junto a la presencia de albinismo oculocutáneo y diátesis hemorrágica son puntos diagnósticos distintivos. El compromiso pulmonar es un carácter diferencial entre el SHP y el síndrome de Chediak-Higashi que además de hipopigmentación y sangrado fácil presenta el antecedente de infecciones recurrentes durante la niñez, neuropatía central y periférica junto a intenso de compromiso de órganos linfoides(17).
La patogenia de la fibrosis pulmonar apunta a la disfunción de las células epiteliales y macrófagos alveolares que, debido al tráfico intracelular distorsionado por orgánulos estresados, sufren apoptosis lo cual promueve la acumulación de fibroblastos con subsecuente depósito excesivo de matriz extracelular(18). El grupo de genes Rab pertenece a la superfamilia de proteínas Ras y los neumocitos tipo II tienen particularmente una expresión muy intensa del Rab38, cuya mutación (BLOC-3) causa homeostasis alterada del surfactante y el desarrollo de cuerpos lamelares gigantes en las células epiteliales que con la presencia de “macrófagos espumosos” (depósitos de lipofuscina ceroide) caracterizan a la FPSHP(19-20). Las opacidades en vidrio esmerilado representan estos acúmulos celulares y distinguen a este síndrome de la FPI, cuya expresión histológica es la neumonitis intersticial usual (UIP). En la FPSHP, sin embargo, pueden verse también neumonitis intersticial no específica (NINE) y neumonitis intersticial descamativa (NID) en el análisis histológico de los tejidos(11). Estas tres entidades tisulares (UIP, NINE y NID) tienen actualmente diferentes propuestas terapéuticas, cuando no asociados a SHP. ¿Debemos extrapolar este enfoque al contexto del síndrome? De lo contrario es tiempo de asumir que la clasificación actual de las neumonías intersticiales representa apenas un espectro estereotipado y reaccional a distintas causas. Nuestro telescopio James Webb se estructura sobre lo que la biología molecular nos contará de las EPID.
Debido a que es una enfermedad infrecuente el relato de la historia natural es inconsistente, con tasas variables de progresión. Se mencionan exacerbaciones agudas de fibrosis pulmonar asociada a rápido empeoramiento de los síntomas y también casos con <20 meses de sobrevida después de alcanzar CVF < 50% del previsto, como hemos comprobado en el trabajo aquí presentado(14,21). Hasta la fecha parece imposible predecir la evolución y mucho menos contener la FPSHP con fármacos, siendo el trasplante pulmonar la única posibilidad terapéutica adjunta a la atención preventiva y de apoyo acorde a grado de disfunción respiratoria (evitar humo de cigarrillo y otros irritantes pulmonares, profilaxis de infecciones con vacunas antivirales y antineumocócica, rehabilitación, y oxigenoterapia suplementaria). Los corticoides no deben utilizarse pues no se han encontrado resultados favorables en pequeños ensayos. En relación a los antifibróticos, recientemente aprobados para el tratamiento de la FPI, existe mayor experiencia con la pirfenidona que con el nintedanib, aunque con resultados no concluyentes en diferentes ensayos(14,22). El horizonte terapéutico se proyecta hacia la transferencia de genes por lentivirus, la edición genética mediante CRISPR-Cas9 u organoides alveolares derivados de células madres pluripotenciales(7).
En conclusión, en este reporte se describen datos epidemiológicos, patogénicos, clínicos y diagnósticos de dos casos de fibrosis pulmonar familiar asociados al síndrome de Hermansky-Pudlak. Deseo enfatizar, in memoriam, la necesidad de iniciar el proceso de implementación del trasplante pulmonar en el Paraguay. En los últimos años se han dado pasos históricos en el seno de la conciencia colectiva y dentro del compromiso gerencial de la ablación y trasplante. Se beneficiarán muchos compatriotas portadores de fibrosis quística, silicosis, EPID, EPOC, hipertensión pulmonar y otras enfermedades respiratorias limitantes.

