











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
En la actualidad, nuestro país carece de medicamentos genéricos que aseguren un tratamiento terapéutico eficaz a la población, solo existen medicamentos similares que no han sido sometidos a estudios de bioequivalencia, por lo que no se podría establecer intercambiabilidad con el medicamento innovador, que fue aquel que salió primero al mercado y que recibió la autorización de comercialización por la demostración de eficacia y seguridad con base en ensayos clínicos1.
La intercambiabilidad se fundamenta en que dos equivalentes farmacéuticos son bioequivalentes, cuando las concentraciones del fármaco se equilibran en el organismo sincrónicamente, por lo tanto, la evolución de las concentraciones en la biofase va a ser prácticamente la misma para ambos productos (multifuente e innovador)2.
La prueba de disolución in vitro es utilizada en el desarrollo y evaluación de medicamentos para proporcionar control del proceso y aseguramiento de la calidad, así como determinar las características de liberación de estabilidad del producto a lo largo del tiempo, y de esta manera cumplir con parámetros de identificación, pureza, y, bioequivalencia en el caso de productos farmacológicos intercambiables3,4.
La bioequivalencia in vitro busca establecer correlaciones in vitro - in vivo, de tal forma que una prueba de disolución pueda reemplazar la disposición del medicamento cuando es administrado en el ser humano, con la finalidad de obtener equivalentes biofarmacéuticos entre un medicamento innovador y multifuente, se utilizan 12 unidades de dosificación y se colocan en medios agitados con diferentes tipos de pH (1,2; 4,5 y 6,8), que simulan las condiciones del tránsito del medicamento desde el inicio de la administración, que se analizan generalmente mediante cromatografía o espectroscopia ultravioleta5-7.
La similitud de los perfiles de disolución para fines de intercambiabilidad se determina mediante el método de modelo independiente del factor de similitud (f2), que debe considerar por lo menos 3 puntos de muestreo para cada medio de disolución, y debe presentar un valor comprendido entre 50 - 100. Asimismo, el ajuste de los porcentajes de la evolución temporal del fármaco se realiza mediante el modelo dependiente, e incluye el orden cero, primer orden, Higuchi, raíz cúbica y Weibull3,8,9.
La prednisona es un glucocorticoide utilizado en la práctica clínica, y se prescribe en enfermedades inflamatorias en forma de tabletas orales de liberación inmediata en presentaciones de 5, 20 y 50 mg, pertenece a la clase 1, de acuerdo al Sistema de Clasificación Biofarmacéutica (SCB), y sólo debe demostrar equivalencia terapéutica in vitro para demostrar su intercambiabilidad entre el medicamento multifuente y referente10,11.
La United States Pharmacopeia (USP) 41 establece como criterio de aceptación un contenido no menor del 90% y no más del 110% de la cantidad declarada para tabletas que declaran contener más de 10 mg de prednisona, y para el ensayo de disolución establece una tolerancia de no menos del 80% (Q) a los 30 minutos, utilizando como medio agua 900 mL, aparato tipo paleta a 50 rpm, en modo ultravioleta a 242 nm de lectura12.
En el Perú, no existen estudios publicados que reporten los perfiles de disolución de prednisona, por lo que, la realización de este tipo de tendría implicancias en los sistemas de salud de la población peruana, al poder tener un producto eficaz y seguro a un precio menor.
El estudio tuvo como propósito comparar los perfiles de disolución de tabletas de prednisona 20 mg comercializados en Perú, y de esta manera poder establecer intercambiabilidad entre la formulación de referencia y multifuente.
MATERIAL Y MÉTODOS
El estudio fue cuantitativo comparativo con diseño no experimental, y se utilizaron 50 tabletas de la formulación referente y multifuente de prednisona 20 mg procedentes del mismo lote y con fecha de vencimiento similar.
Las curvas de calibración fueron preparadas a partir de un estándar secundario de prednisona para los tres medios de disolución (buffer pH 1,2; buffer pH 4,5 y buffer pH 6,8). Se preparó el estándar de prednisona mediante un estudio piloto. Se pesaron 25 mg de estándar de prednisona y se colocaron en un matraz aforado de 250 mL y se procedió a aforar. Finalmente se filtraron las muestras y se realizaron lecturas a una longitud de onda de 242 nm.
El ensayo de disolución para los tres medios utilizó doce tabletas para cada formulación referencia y multifuente, y se siguieron los lineamientos establecidos por la USP 40: aparato tipo II, 50 rpm, volumen de agua 900 mL, temperatura 37 ºC±0,5 ºC. Se tomaron alícuotas filtradas de 10 mL sin reposición en diferentes tiempos (5, 10, 15, 30, 45 y 60 minutos). Posteriormente se realizaron lecturas en el espectrofotómetro ultravioleta a 242 nm. Las concentraciones se determinaron a partir de las curvas estándares previamente elaboradas12.
Los modelos dependientes permitieron establecer el mejor ajuste de los datos de los porcentajes de fármaco disuelto, mediante el Criterio de Información Akaike (AIC), y a partir del mejor modelo cinético para cada lote y pH se determinaron sus constantes de velocidad de disolución13-15.
Los modelos independientes se establecieron mediante el cálculo de f215, y según lo establecido por la Food and Drug Administration (FDA) que sugiere que dos perfiles de disolución se considerarán similares si el valor se sitúa entre 50 y 1007.
El tiempo medio de disolución (TMD) se calcula a partir de las curvas acumulativas de las cantidades disueltas de fármaco en función al tiempo mediante la ecuación, y es el tiempo promedio de residencia del principio activo en la forma farmacéutica. La eficiencia de disolución (ED%) se calcula a partir de los valores obtenidos del área bajo la curva (ABC) del perfil de disolución para cada intervalo de tiempo, a través del método de los trapezoides13.
Los parámetros evaluados fueron caracterizados mediante estadísticos descriptivos, media aritmética y coeficiente de variación porcentual. Los promedios de las constantes de disolución para cada formulación y para cada pH fueron sujetos a un análisis t de Student con un nivel de confianza del 5% (( = 0,05)5,6.
RESULTADOS
En la Figura 1 se reportan los porcentajes de disolución de las tabletas de prednisona 20 mg hasta los 60 minutos a buffer pH 1,2; evidenciando una similitud en los perfiles del producto referente y multifuente, en los primeros puntos de muestreo los porcentajes son pequeños y se van incrementando en función del tiempo hasta que finalmente se vuelve constante.

En la Figura 2, a buffer pH 4,5 los perfiles de disolución presentan similitudes en los porcentajes disueltos, lo que evidenciaría una primera fase de liberación con incremento en los coeficientes de variación porcentual, y a medida que se incrementa el tiempo se observa una mayor cantidad de fármaco disuelto con un menor coeficiente de variación porcentual.

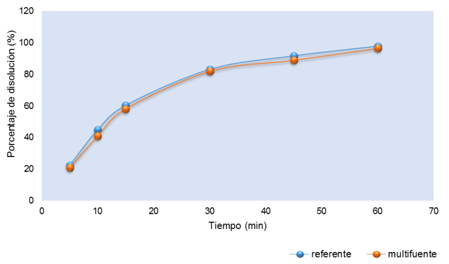
En la Figura 3, la tendencia de los perfiles de disolución en función del tiempo a buffer pH 6,8 se hacen constantes a los 60 minutos con similitudes en los porcentajes disueltos, lo que demostraría el compromiso que presentan las industrias farmacéuticas peruanas en el proceso de manufactura de sus medicamentos, utilizando principios activos y excipientes con las mismas exigencias que los laboratorios transnacionales, asegurando eficacia terapéutica en el paciente.
En la Tabla 1 se puede observar que los porcentajes promedio de fármaco disuelto en función del tiempo siguen una cinética de Weibull según el Criterio de Información de Akaike, el cual evalúa el valor más pequeño en los pH 1,2; 4,5 y 6,8 tanto para referente y multifuente.
Tabla 1: Criterio de Información de Akaike para la cinética de disolución de las tabletas de prednisona 20 mg referente y multifuente a pH 1,2; 4,5 y 6,8.
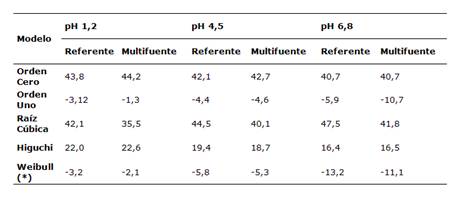
(*) Modelo que explica la cinética de disolución de prednisona en tabletas en los tres medios de disolución.
En la Tabla 2 se analiza estadísticamente el tiempo medio de disolución de las tabletas de prednisona 20 mg referente y multifuente a pH 1,2; 4,5 y 6,8; para lo cual se realizó en primer lugar la normalidad de los datos mediante la prueba Shapiro Wilk; posteriormente, se evaluó diferencia significativa mediante la prueba t student, y se encontró que en los tres medios de disolución no es significativo, por lo que se asumiría igualdad en las medias para el producto referente y multifuente.
Tabla 2: Tiempo medio de disolución de las tabletas de prednisona 20 mg referente y multifuente a pH 1,2; 4,5 y 6,8.

(*) El valor es significativo a nivel de 0,05 y grados de libertad 22.
De la misma manera, en la tabla 3 se analiza la eficiencia de disolución de las tabletas de prednisona 20 mg referente y multifuente a pH 1,2; 4,5 y 6,8; habiendo comprobado en primer término la normalidad de los datos con la prueba de Shapiro wilk, y finalmente no se encontró diferencia estadísticamente significativa en los tres medios de disolución mediante la prueba t de student. En este sentido, se asumiría igualdad entre el producto referente y multifuente.
Tabla 3: Eficiencia de disolución de las tabletas de prednisona 20 mg referente y multifuente a pH 1,2; 4,5 y 6,8.
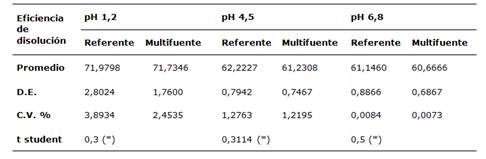
(*) El valor es significativo a nivel de 0,05 y grados de libertad 22
En la Tabla 4 se aprecia el cálculo del factor de similitud (f2), evidenciando que en los tres medios de disolución alcanzó valores mayores a 50, y según la normatividad establecida por la Food and Drug Administration, se establecería similitud en los perfiles de disolución por esta comprendido en el rango de 50 - 100, así como presentar al menos 4 puntos de muestreo, y presentar coeficiente de variación porcentual menores al 20% los primeros puntos y 10% en los últimos puntos, por tanto, se podría inferir intercambiabilidad entre las formulaciones referente y multifuente de tabletas de prednisona 20 mg comercializadas en el Perú.

DISCUSIÓN
Los perfiles de disolución de los productos referente y multifuente en los buffers pH 1,2; 4,5 y 6,8; y según el protocolo de estudio de bioequivalencia in vitro establecida por la Organización Mundial de la Salud (OMS), los buffers estarían representando condiciones fisiológicas, simulando así el fluido gástrico, duodenal e intestinal respectivamente. Se aprecia el comportamiento de disolución del producto multifuente en relación al producto de referencia, los cuales mostraron un perfil característico, correspondiente a una completa disolución y a una rápida velocidad10.
La United State Pharmacopeia 41 indica que no menos del 80% de la cantidad declarada del producto con principio activo de prednisona 20 mg se disuelve en 30 minutos; y como se evidencia ambas formulaciones cumplen con lo establecido: minuto 30 a pH 1,2 (referente: 85,6 % y multifuente: 83,3 %), minuto 30 a pH 4,5 (referente: 84,1% y multifuente: 83,2 %) y minuto 30 pH 6,8 (referente: 83,1 % y multifuente: 81,9 %)12.
La cinética de disolución de las dos formulaciones en estudio y para cada pH se realizó con el Criterio de Información de Akaike, que presentó modelos dependientes como orden cero, primer orden, raíz cúbica, Higuchi y Weibull13. Este método consiste en seleccionar el modelo que minimiza la probabilidad negativa restringida por el número de parámetros, es decir el valor más pequeño señala estadísticamente el mejor proceso de disolución; específicamente tiene como objetivo encontrar el mejor modelo de aproximación para el verdadero proceso de generación de datos desconocidos. En este sentido, el mejor modelo dependiente para ambas formulaciones en los pH 1,2; 4,5 y 6,8 fue Weibull, lo que indicaría que este modelo es el que estadísticamente explica mejor el proceso de disolución en cada medio señalado, y reflejaría la influencia que pueden tener las condiciones de disolución como medio de disolución y pH, tipo de equipo y velocidad de agitación, no solo sobre la velocidad de disolución, sino también sobre la cinética de liberación de los fármacos (8,14,15.
Del mismo modo, el tratamiento estadístico del tiempo medio de disolución en una distribución t de student bilateral con una α=0,05 y 22 grados de libertad; los valores obtenidos fueron: -1,0 a pH 1,2; -0,4 a pH 4,5; -1,2 a pH 6,8. En los tres pH se obtuvieron valores mayores a p>0,05, lo que indicaría que el tiempo medio de disolución del producto multifuente es similar al producto referente. Los resultados evidencian que los excipientes no estarían afectando la velocidad y la extensión de la absorción, aspecto considerado por la Food and Drug administration en el momento que se desea obtener la exención de estudios de bioequivalencia8.
Respecto al tratamiento estadístico de la eficiencia de disolución, este parámetro expresa la disolución del fármaco, y relaciona el área bajo la curva del perfil de disolución del producto en estudio respecto al área total del rectángulo formado por la disolución teórica del 100% de la dosis y el intervalo de tiempo de la prueba (9. La distribución t de student bilateral con un α=0,05 y 22 grados de libertad; arrojó valores de 0,2 a pH 1,2; 0,3 a pH 4,5; 0,5 a pH 6,8; y los valores de p fueron 0,800 a pH 1,2; 0,758 a pH 4,5; 0,5830 a pH 6,8; estadísticamente no significativos, por lo que se podría inferir similitud entre el producto multifuente y referente.
El factor de similitud (f2) en los tres medios de disolución, arrojó valores de 63,9 a pH 1,2; 59,5 a pH 4,5 y 69,9 a pH 6,8. El f2 es una transformación logarítmica del recíproco de la raíz cuadrada de la suma de los cuadrados de los errores, y es una medida de la semejanza en el porcentaje de disolución entre dos curvas, y en la actualidad es adoptado por la Food and Drug Administration y European Agency for the Evaluations of Medicinal Products que establecen la utilización de los porcentajes temporales disueltos de los medicamentos y según la comparación matemática de los perfiles de disolución, cuando presenta valores comprendidos entre 50 -100 indica que hay similitud en las curvas; y por lo tanto es similar el rendimiento del producto multifuente con el producto referente, es decir son productos intercambiables13,14,15.
En el buffer pH 6,8 se obtuvo el valor más alto del factor de similitud (f2) (69,9), y reflejaría que medicamentos como prednisona que tiene un pH de 2,6 - 3,6 se disolvería más fácilmente en un medio alcalino como el pH 6,8. De este modo, pequeñas modificaciones del pH del medio de disolución pueden hacer variar la solubilidad de una determinada sustancia, aumentar o disminuir la fracción ionizada, que es la más soluble en agua, y con ello las implicancias respecto a la absorción de medicamentos14,15.
A nivel nacional e internacional, son pocas las investigaciones realizadas en prednisona, es así, que, dentro de los estudios más recientes y relevantes, Henrique, Deris y Antunes realizaron un estudio en Brasil sobre los perfiles de disolución de prednisona, encontrando que los f2 en los medios pH 1,2; 4,5 y 6,8 fueron mayores a 50, con lo cual, se estaría asegurando que la población brasileña acceda a medicamentos con menor precio11.
Finalmente, el estudio permitió inferir que las tabletas de prednisona 20 mg referente y multifuente comercializados en Perú son similares, por lo que se establecería, intercambiabilidad entre ambas formulaciones, con base en pruebas de perfiles de disolución in vitro, y de esta manera tener alternativas farmacéuticas para el tratamiento de patologías de carácter inflamatorio, cumpliendo con los objetivos estratégicos que presenta el Ministerio de Salud, de que la población, específicamente, aquellos con estratos socioeconómicos bajos, puedan acceder a medicamentos de calidad. Asimismo, los resultados del presente estudio, deberían complementarse con un estudio in vivo, para obtener datos que midan la magnitud (área bajo la curva) y la velocidad de absorción (concentración máxima y tiempo máximo).

