










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
On-line version ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud vol.14 no.1 Asunción Apr. 2016
https://doi.org/10.18004/Mem.iics/1812-9528/2016.014(01)94-109
Articulo Revisión/ Review Article
Manifestaciones clínicas y laboratoriales en el Lupus Eritematoso Sistémico-LES
Clinical and laboratory manifstations in Systemic Lupus Erythematosus
Isabel Acosta ColmánI, Gabriela AvilaI, Maria Eugenia AcostaI, Alicia AquinoI, *Osmar CenturiónII, Margarita DuarteI
I Departamento de Reumatología. Hospital de Clínicas. Universidad Nacional de Asunción
II Departamento de Medicina Interna. Hospital de Clínicas. Universidad Nacional de Asunción
R E S U M E N
El Lupus eritematoso sistémico (LES) es una enfermedad autoinmune compleja que se caracteriza por su capacidad de afectar a diversos órganos, lo que determina las diferentes manifestaciones clínicas objetivadas durante la evolución de la enfermedad. De forma asociada se ha descrito que estas manifestaciones presentan una variación geográfica o étnica, siendo por lo general menos grave en pacientes con ascendencia europea que en aquellos que presentan ascendencia africana, asiática o hispana. Alteraciones, tanto del sistema inmune adaptativo (células T y B) como del innato (Toll like receptorx-TLR), contribuyen al desarrollo del LES. Las células B tienen su papel en la producción de los autoanticuerpos (i.e. anticuerpos anti-ADN y anticuerpos anti-nucleosoma) y de determiandas citocinas. Las pruebas de laboratorio son de gran valor cuando se evalúa a un paciente con sospecha de enfermedad autoinmune. Los resultados pueden confirmar el diagnóstico, estimar la severidad de la enfermedad, evaluar el pronóstico y son de suma utilidad para el seguimiento de la actividad del LES.
Palabras clave: manifestaciones clínicas, lupus eritematoso sistémico, análisis laboratoriales.
A B S T R A T
Systemic Lupus Erythematosus (SLE) is a complex autoimmnune disease characterized by its ability to affect different organs, which determines different clinical manifestations observed during the course of the disease. It has been described that these manifestations have geographic or ethnic varations being generally less serious in patients of European descent than in those with African, Asian or Hispanic descents. Alterations of both the adaptative (T and B cells) and innate (Toll like receptorx-TLR) immnune systems contribute to the development of SLE. B cells have a role in the production of auto-antibodies (i.e. anti-DNA and anti-nucleosome antibodies) and some cytokines. Laboratory tests are invaluable when evaluating a patient with suspected autoimmune disease. The results can confirm the diagnosis, estimate the severity of the disease, assess prognosis and are extremely useful for monitoring the activity of SLE.
Keywords: clinical manifestations, systemic lupus erythematosus, laboratory tests.
INTRODUCCIÓN
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune de etiología desconocida, que se caracteriza por la gran variedad de manifestaciones clínicas que pueden presentarse durante la evolución de la enfermedad.
La estimación de la incidencia y prevalencia del LES es variable según las diferentes series. Esto estaría explicado por el diferente número de pacientes incluidos en cada estudio, la metodología de recolección de la información, las fluctuaciones temporales así como también las diferencias regionales (1). Estudios recientes han objetivado una incidencia variable, con valores que varían entre 1,0 por 100.000 en población danesa a valores de 8,7 por 100.000 en Brasil (1). Tal y como se ha mencionado previamente, se ha constatado que esta incidencia no es estática, si no que presenta fluctuaciones cíclicas a lo largo del tiempo. En este sentido, un estudio realizado en los Estados Unidos de América, ha objetivado un aumento de 3 a 7 veces en la incidencia entre los años 1950 y 1992 (1).
Se considera que el lupus es una enfermedad compleja de etiología desconocida en la que están implicados tanto factores genéticos como ambientales (i.e. genero, edad, hormonas, tabaquismo, infecciones, drogas y las anormalidades tanto de la inmunidad innata como en la adaptativa) (2). En los últimos años se ha observado un interés creciente por el estudio genético de esta enfermedad. El avance en las técnicas de genotipado y de los soportes informáticos ha permitido identificar y localizar un elevado número de marcadores genéticos de susceptibilidad para el desarrollo del LES, favoreciendo con esto un mayor conocimiento de esta entidad (3,4).
Tal y como se ha mencionado previamente, las manifestaciones clínicas en el LES son extremadamente heterogeneas, pudiendo afectar a varios órganos. Puede afectar a la piel o las membranas mucosas, las articulaciones, el cerebro, el corazón, el riñón, el pulmón y ocasionalmente el tracto gastrointestinal (5-7). De forma interesante, se ha objetivado que la frecuencia con que se presentan las diferentes manifestaciones clínicas muestra una variación geográfica y étnica importante. Así pues, los pacientes con ascendencia europea presentan manifestaciones cutáneas con más frecuencia que los pacientes de otras zonas. En este sentido se ha descrito que los pacientes con ascendencia africana presentan enfermedad renal con mayor frecuencia que sus pares europeos (1).
En relación al pronóstico, varios estudios han demostrado que determinadas características clínicas como la etnia, el género, la edad, y la condición socioeconómica (i.e. ingreso económico, el nivel de educación y el acceso a los servicios de salud) son variables importantes asociadas con el pronóstico del LES (8-12). Las personas de raza negra tienen una incidencia y prevalencia tres veces mayor y desarrollan el LES en forma más temprana que la raza blanca (13, 14). Se ha observado que los hispanos desarrollan una enfermedad más severa y con una peor evolución que los pacientes caucásicos (15-17).
Las pruebas de laboratorio son de gran valor cuando se evalúa a un paciente con sospecha de enfermedad autoinmune. Los resultados pueden confirmar el diagnóstico, estimar la severidad de la enfermedad, evaluar el pronóstico y son de suma utilidad para el seguimiento de la actividad del LES (18). El objetivo del presente manuscrito fue realizar una revisión de la fisiopatología, las manifestaciones clínicas y laboratoriales del objetivadas en el LES. Un mayor conocimiento de esta desafiante enfermedad nos permitirá realizar un mejor diagnóstico y manejo de los pacientes.
Mecanismos inmunológicos en el Lupus Eritematoso Sistémico
Los mecanismos implicados en la patogenia del LES han sido estudiados intensamente, pero a pesar de este esfuerzo, aun no se ha conseguido conocer el mecanismo exacto implicado en el desarrollo de la enfermedad. Estudios previos han objetivado una producción excesiva de autoanticuerpos, formación de complejos inmunes y daño tisular inmunológicamente mediado. En estos estudios se ha objetivado que la inflamación observada en los diferentes órganos, se produce por el depósito de los complejos inmunes producidos por la union de los autoanticuerpos con los autoantígenos (19).
La disregurlacción de la respuesta inmune innata desempeña un papel importante en la patogénesis del LES. Esta contribuye tanto a la lesión tisular mediante la liberación de citoquinas inflamatorias, así como a la activación aberrante de las células T y B autorreactivas. Esto a su vez ocasiona un incremento en la producción de autoanticuerpos con la resultante lesión orgánica (19-22).
Se considera que la producción aumentada de estos autoanticuerpos es secundaria a una mayor cantidad de nucleosomas. Este aumento en el número de nucleosomas refleja una apoptosis acelerada además de un aclaramiento defectuoso de citocinas (21,23). Todo esto sumado a la deficiencia en la función fagocítica mononuclear y del clearence de los complejos inmunes, hace que el LES sea utilizado como un modelo de estudio para la comprensión de la inmunopatogénesis de las enfermedades mediadas por complejos inmunes.
Factores asociados al desarrollo del LES
Como se ha citado previamente, el LES se caracteriza por una pérdida global de la auto-tolerancia. Se considera que esta con la disregulación inmune posterior, son consecuencia de la interacción de determinados desencadenantes ambientales con factores genéticos (22). Los genes del Complejo Mayor de la Histocompatibilidad (CMH), particularmente el HLA1, B8, y DR3 han sido relacionados con la patogenia del LES (24-26). Los genes del CMH están asociados a un riesgo aumentado de una respuesta autoinmune para autoantígenos y por lo tanto a un riesgo para las enfermedades como el LES (25,26). Además de la asociación con el locus del HLA-DR, se han identificado otras asociaciones como la del STAT 4 (signal transducer and activator of transcription 4), PTPN 22 (protein tyrosine phosphatase nonreceptor type 22), ITGAM (integrin alpha M) entre otros (27-30).
No se conoce con exactitud cuales son los elementos exógenos que al interactuar con un individuo genéticamente predispuesto desencadenaran la enfermedad. Se han postulado diversos factores, como el género, determinadas drogas, infecciones virales y la exposición a la radición ultravioleta.
Teniendo en cuenta que el 90% de los pacientes con lupus son mujeres, varios estudios han analizado el papel de las hormonas femeninas o un efecto de los genes contenidos en el cromosoma X en el desarrollo del LES (31,32).
Varias drogas pueden causar una variante del Lupus llamado Lupus inducido por drogas. Las drogas mejor conocidas son la procainamida, hidralazina y quinidina. Se ha descrito que los pacientes con lupus inducido por drogas presentan manifestaciones dérmicas y articulares; siendo las características renales y neurológicas menos frecuentes (19,31,33,34).
Los antecedentes de enfermedades virales pueden estar presentes al inicio del lupus o inmediatamente antes de una recaída. El virus del Epstein Barr (EBV) puede ser importante ya que una asociación temporal entre el inicio del Lupus y la ocurrencia de una infección por el EBV ha sido reportada.
Finalmente, la radiación ultravioleta es el factor ambiental mejor ligado al Lupus. Tal es la importancia en el desarrollo del LES que la fotosensibilidad fue considerada un criterio del Colegio Americano de Reumatología para la clasificación de la enfermedad (35,36).
Manifestaciones clínicas del Lupus Eritematoso Sistémico
El LES al ser una enfermedad inflamatoria multisistémica, se caracteriza por la capacidad de afectar a varios órganos. Así pues, se puede objetivar afectación de la piel o las membranas mucosas, las articulaciones, el cerebro, el corazón, el riñón, el pulmón y ocasionalmente el tracto gastrointestinal (37).
Los síntomas generales como la fatiga, el malestar general, la fiebre, la anorexia, y la pérdida de peso son hallados con elevada frecuencia; tanto como síntomas iniciales de la enfermedad o como complicaciones de ésta. La fatiga merece una mención especial debido a que es un síntoma muy incapacitante que está ligado a un trastorno depresivo y se presenta de forma independiente de las manifestaciones clínicas o serológicas (38-40). Por otro lado cabe mencionar que la valoración del sindorme febril es un verdadero desafío en este grupo de pacientes. Se ha descrito que puede presentarse en hasta el 42% de los pacientes como una manifestación de la actividad inflamatoria. Pero ante este síntoma, siempre deben ser descartadas otras causas de la fiebre, como la presencia de cuadros infecciosos intercurrente, tumores malignos y el efecto de determinadas drogas (39).
Manifestaciones cutáneas
La afectación cutánea es amplia y variable, por lo que identifican manifestaciones específicas y no especificas de la enfermedad. Actualmente se consideran como manifestaciones específicas del LES al Lupus cutáneo agudo, subagudo y crónico tal y como se observa en la Tabla 1 (41).
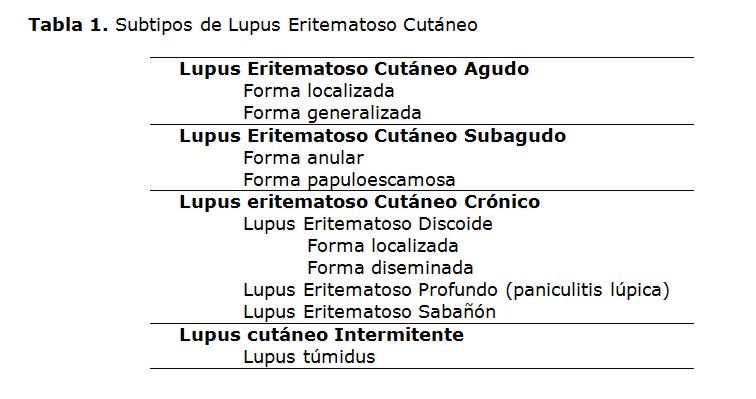
Las lesiones agudas del LES se presentan con el clásico rash malar o en ala de mariposa, el cual puede afectar solo la piel transitoriamente, precediendo al inicio de la enfermedad multisistémica (42-44). Las lesiones agudas generalizadas son menos comunes y su inicio usualmente coincide con una exacerbación de la enfermedad sistémica orgánica y/o una actividad de la enfermedad prolongada. Se caracterizan por un eritema maculopapular de distribución simétrica, que en ocasiones se asocia a prurito (45).
Las lesiones subagudas presentan una distribución simétrica en las áreas fotoexpuestas siendo inicialmente máculas o pápulas eritematosas que posteriormente se vuelven placas anulares/policíclicas o papuloescamosas. Estas lesiones también pueden ser desencadenadas por la luz ultravioleta o por diferentes drogas (i.e. antifúngicos, diuréticos, bloqueadores de los canales de calcio, etc) (46).
En relación al lupus cutáneo crónico, éste se manifiesta de tres formas diferentes; el lupus discoide, la paniculitis lúpica/lupus profundus y el lupus sabañón. El lupus discoide es la forma más frecuente y puede presentarse tanto de forma localizada (i.e. cara, orejas) o como una forma diseminada cuando compromete por debajo del cuello (47). La paniculitis lúpica es menos frecuente y se presenta como nódulos indurados o placas que resultan de una lipoatrofia profunda (43,44). El lupus sabañón se caracteriza por presentar placas púrpuricas dolorosas localizadas en zonas acras expuestas al frío (48).
El lupus eritematoso túmido ha sido distinguido como una entidad distinta del lupus cutáneo crónico. Clínicamente se caracteriza por presentar placas semejantes a la urticaria. Estas lesiones se localizan en zonas expuestas al sol y se caracterizan por ser induradas, únicas y con una superficie rojiza o violácea. A diferencia de la variedad discoide, el lupus tumidos rara vez progresa a enfermedad sistémica (49-51) .
Las manifestaciones cutáneas no específicas abarcan un gran número de manifestaciones que tal y como su nombre lo indica, pueden estar asociadas a otras enfermedades. Entre ellas se incluyen la telangiectasia periungueal, la lívedo racemosa, la tromboflebitis, el fenómeno de Raynaud y la vasculopatía oclusiva acral (41).
Manifestaciones articulares
El compromiso articular es una de las primeras manifestaciones de la enfermedad. Esta se puede presentar como una artropatía deformante no erosiva o artropatía de Jacoud;
artritis deformante simétrica erosiva, similar a la artritis reumatoide y como una artritis no deformante (39,52).
La artropatía de Jacoud se ha descrito en el 10 a 35% de los pacientes. Es una condición capsular y periarticular generalizada que puede afectar a todas las articulaciones en especial las de las manos. Esta artropatía es secundaria a la laxitud de la capsula, tendones y ligamentos que a su vez ocasionan inestabilidad de la articulación. Si el proceso inflamatorio se localiza en las pequeñas articulaciones de las manos y este no es controlado, puede observarse rigidez de las mismas debido a la fibrosis de la capsula articular y de los ligamentos afectos (52). Los pacientes que presentan artritis erosiva poseen manifestaciones articulares similares a la artritis reumatoide además de síntomas de LES más leves. Las erosiones pueden ser secundarias a la sinovitis destructiva, fricción con tendones inflamados y a las fuerzas mecánicas alteradas resultantes de la subluxación (52).
Es importane recordar que los pacientes con LES pueden presentar mialgias. Esta puede ser una manifestación propia de la enfermedad, ser secundaria a la artritis, al uso de determinados fármacos (i.e. corticoesteoroides), o de una miositis. Se ha descrito que los pacientes con miositis presentan más alopecia, lesiones de las mucosas, artritis erosiva y síndrome seco (52).
Manifestaciones hematológicas
Este tipo de manifestaciones, es frecuentemente objetivada en los pacientes con LES. De las posibles citopenias, la anemia es la manifestación hematológica más frecuente, y ha sido descrita en el 50 al 78% de los pacientes. Puede observarse anemia secundaria a enfermedades crónicas, por ferropenia y la hemolítica autoinmune (39,53) . La anemia de enfermedades crónicas es la más comúnmente identificada en los pacientes lúpicos. Esta anemia, en la mayoría de los casos, es moderada.
La anemia ferropénica es por frecuencia la segunda causa de anemia en el LES y puede ser secundaria a metrorragia o a perdidas gastrointestinales. La gravedad de la misma generalmente se correlaciona con el grado de la actividad de la enfermedad y con un nivel elevado de eritropoyetina. A diferencia de esta, la anemia por enfermedades crónicas y la anemia hemolítica autoinmune se caracterizan por niveles inadecuados de eritropoyetina (54). Es importante rescatar que en las anemias autoinmunes, el test de Coombs es positivo en solo el 10% de los pacientes con hemolisis clínicamente significativa (40).
La neutropenia en el LES generalmente es secundaria al uso de las drogas inmunosupresoras, si bien también puede objetivarse anticuerpos que inhiben las colonias de crecimiento de granulocitos en la médula ósea. En relación a la trombocitopenia, se distinguen dos subtipos diferentes, un subgrupo en el cual la trombocitopenia es secundaria a la activación multisistémica severa y un segundo subgrupo en el cual la trombocitopenia es un hallazgo aislado (38,40) .
Se ha descrito que en ocasiones, la trombocitopenia puede ser parte de una púrpura trombótica trombocitopénica (PTT) y empeorando el pronóstico del paciente con LES (53, 54) .
Manifestaciones neuropsiquiátricas
Tanto el Sistema Nervioso Central (SNC) como el sistema nervioso periférico (SNP) pueden estar comprometidos en el LES. Como las manifestaciones pueden ser tanto neurológicas como psicológicas, comúnmente son llamadas neuropsiquiátricas. El compromiso del SNC puede ser difuso y manifestarse como disfunción cognitiva, trastornos del humor y psicosis; o ser focal y presentarse como accidentes cerebrovasculares (55).
La afectación neuropsiquíatrica en el LES se ha descrito en más del 50% de lo pacientes, siendo la disfunción cognitiva, la manifestación más frecuentemente reportada (56-58) . El LES puede afectar todos los dominios cognitivos asociándose frecuentemente a una discapacidad social y dishabilidad funcional (59). Otras anormalidades neuroanatómicas observadas en los pacientes con esta manifestación son la atrofia cortical, los infartos mayores, la desmielinización en parches, mientras que la vasculitis pura es rara (60-62) .
En los pacientes con este tipo de manifestación, se ha objetivado daño de células neuronales y gliales, así como un incremento en la proteína ácida neurofibrilar glial y de la proteína triple del neurofilamento. La presencia de pleocitosis y de bandas de inmunoglobulina oligoclonal en el LCR son otras anormalidades comunes que reflejan una activación intratecal del sistema inmune (<">63-65) . En los estudios de imagen como la RMN cerebral se observa una atrofia e hiperintensidad de la sustancia blanca (66,67) . En los últimos años se ha observado un papel patogénico de los anticuerpos anti proteína ribosomal P que reconocen proteínas específicas en los ribosomas (67,68) .
Manifestaciones renales
El compromiso renal es una de las manifestaciones más graves del LES, con una elevada morbilmortalidad. El compromiso renal del LES abarca desde una nefritis silente considerada como el estadío más temprano del compromiso renal a un síndrome nefrótico con deterioro del filtrado glomerular. Este último puede presentar una progresión rápida a enfermedad renal terminal, lo que determina la necesidad de un diagnóstico temprano. Las características más comúnmente encontradas en la nefritis lúpica son la proteinuria, presencia de cilindros urinarios, hematuria, piuria, aumento del nivel de creatinina e hipertensión (69) .
La biopsia renal es esencial para determinar el tipo de compromiso y esto es fundamental para su manejo terapéutico y pronóstico. La mayoría de los pacientes presentan alguna anomalía en la biopsia renal manifestándose esto en la microscopía de luz o con otras técnicas especiales como la inmunofluorescencia o la microscopía electrónica.
La afectación renal puede ser secundaria a una glomerulonefritis, a nefritis intersticial, enfermedad tubular, microangiopatía trombótica, vasculitis, aterosclerosis o vasculopatía lúpica. La clasificación de nefropatía lúpica de la Sociedad Internacional de Nefrología y de la Sociedad de Patología Renal consideran seis tipos de afectación renal (Tabla 2) (70-72).
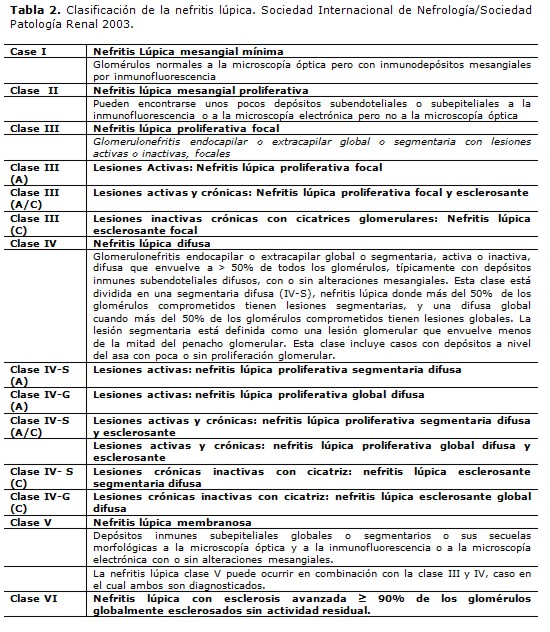
La remisión completa de la nefritis lúpica se ha asociado a una supervivencia a los 10 años de un 92%. A diferencia de lo citado para la remisión completa, solo un 43 a 45% de los pacientes con remisión parcial sobreviven a los 10 años (69,71,72). Las recaídas renales en la nefritis lúpica ocurren de un 27% a un 66%; y esto se asocia a un mayor riesgo de deterioro renal posterior con toxicidad adicional por las drogas inmunosupresoras (73).
Manifestaciones gatrointestinales
Cualquier área del aparato gastrointestinal puede estar comprometido en el LES, pudiéndose presentar como una enfermedad esofágica, una vasculitis mesentérica, una enfermedad inflamatoria intestinal, una pancreatitis, una enfermedad hepática o una peritonitis (74). La disfagia se objetiva en un pequeño porcentaje de los pacientes y puede estar asociada tanto a odinofagia, como también a alteraciones del ritmo peristáltico esofágico. Esta última manifestación se observa en los pacientes que presentan fenómeno de Raynaud (74). Los pacientes con LES pueden tener compromiso de los vasos mesentéricos por vasculitis o trombosis (75).
La pancreatitis aguda se ha descrito en el 5-10% de los pacientes con LES y la mayoría de estos tienen una enfermedad activa en el momento de la presentación de la misma. Se ha descrito que la mortalidad de la pancreatitis lúpica, es de un 27% mayor a la observada en la pancreatitis no asociada a esta enfermedad (76).
El compromiso hepático puede llevar a una enfermedad grave como por ejemplo cirrosis hepática, hepatitis crónica activa, hepatitis granulomatosa, hepatitis crónica persistente y esteatosis; siendo esta última la lesión más común observada en el LES. Todas estas lesiones pueden ser secundarias al LES o a las drogas hepatotóxicas prescriptas para el tratamiento del LES (77,78) . La peritonitis constituye cerca del 30% de todos los episodios de serositis en el LES. La ascitis puede estar asociada en el 11% de los casos con peritonitis; sin embargo en los estudios de autopsia ha sido hallada hasta en un 60% de los casos (74).
Manifestaciones pulmonares
La patología pleuropulmonar puede ser una complicación importante en el LES; aunque la mayoría de los pacientes no desarrollan una intersticiopatía clínicamente evidente, el LES como patología autoinmune es responsable del 2% de las muertes provocadas por una afectación pulmonar (79). Las manifestaciones pleuropulmonares del LES incluyen a la pleuritis lúpica; neumonitis aguda o crónica, hemorragia pulmonar, embolismo pulmonar e hipertensión pulmonar. Las manifestaciones pleurales han sido reportadas en el 30-60% de los pacientes con LES, si bien en las autopsias de pacientes lúpicos se ha encontrado hasta un 93% de compromiso pleural. Los derrames pleurales generalmente son bilaterales y en pequeña cantidad con características de exudados (80).
La neumonitis puede tener una presentación aguda o crónica. La primera ocurre generalmente como una recaída de la enfermedad multisistémica; los pacientes se presentan con tos, disnea, dolor torácico, fiebre e inclusive hemoptisis. La radiografía y la tomografía axial computarizada, muestran usualmente un infiltrado intersticial bilateral en vidrio esmerilado. La neumonitis lúpica crónica generalmente se presenta como una enfermedad pulmonar intersticial y se caracteriza por tos no productiva, disnea de esfuerzo, fatiga y rales basales (80). La hemorragia alveolar es una complicación seria, con una mortalidad entre el 50-90%. Se presenta característicamente con disnea de inicio brusco, tos, fiebre, infiltrados pulmonares y hemoptisis en un 50% de los casos produciendo una caída brusca de la hemoglobina.
La hipertensión pulmonar (HTP) puede ser secundaria a la actividad de la enfermedad o a complicaciones como el embolismo pulmonar, enfermedad valvular cardiaca o enfermedad pulmonar intersticial. Está presente en el 5-14% de los pacientes (81).
Manifestaciones cardiacas
Todas las estructuras cardiacas pueden estar afectadas en los pacientes con LES, desde el pericardio, el miocardio, el endocardio, las arterias coronarias hasta el tejido de conducción (82-84). Este compromiso puede ser secundario a la actividad inflamatoria del LES o a otra enfermedad sistémica como la hipertensión arterial. Algunos estudios de necropsia han demostrado un 40–70% de miocarditis; mientras que la miocarditis sintomática es reportada en solo un 5-10% de los pacientes. Esto pone de manifiesto que el compromiso subclínico cardiaco es un hallazgo común en los pacientes con LES (85-87).
La afectación cardiovascular es una de las causas más importantes de morbilidad y mortalidad en el LES. Manzi et al. (86), han reportado que los eventos coronarios fueron 50 veces más frecuentes para los pacientes con LES en el rango etario de 35–44 años en relación a la población sana de la cohorte de Framingham. En la cohorte de LES del Hospital John Hopkins, el riesgo de presentar un evento cardiovascular fue 2,6 veces mayor que para la población de Framingham incluso después de controlar los factores de riesgo para enfermedad cardiovascular tradicionales en este grupo (87,88).
Diagnóstico y laboratorio en el lupus eritematoso sistémico
Las pruebas de laboratorio son de gran utilidad cuando se evalúa a un paciente con sospecha de enfermedad autoinmune. Los resultados pueden confirmar el diagnóstico, estimar la severidad de la enfermedad, evaluar el pronóstico y son de utilidad para el seguimiento de la actividad de la enfermedad (89).
Los estudios de coagulación como la prolongación del tiempo parcial de tromboplastina activada, el tiempo de protrombina o ambos sugieren la presencia de un factor inhibidor de la coagulación que puede estar presente en el síndrome antifosfolípido secundario al LES. Un incremento en el nivel de las enzimas musculares puede ser visto en el caso de que éste presente miopatía (89). El análisis de orina es comúnmente utilizado para la evaluación del daño renal (glomerulonefritis y nefritis intersticial) donde se puede objetivar proteinuria, hematuria o un sedimento urinario activo (cilindros leucocitarios o de glóbulos rojos) (90). En relación a las proteínas séricas en la respuesta a la inflamación, estas son producidas por el hígado en respuesta al estrés y a la actividad inflamatoria asociada. Las citoquinas proinflamatorias, como IL-1, IL-6 y TNF-α inducen la síntesis de algunos reactantes de fase aguda como la proteína C reactiva (PCR), fibrinógeno y haptoglobina que frecuentemente se encuentran elevados en el LES (91).
Los anticuerpos antinucleares (ANA) son inmunoglobulinas que reaccionan contra diferentes componentes autólogos nucleares y citoplásmicos (92). Se han descrito tres tipos de ANA circulantes. Uno de ellos está presente en todos los individuos a títulos relativamente bajos y son los llamados ANA naturales (92, 93). Un segundo grupo de ANA son los que se producen como resultado de procesos infecciosos; estos no se asocian a manifestaciones clínicas de enfermedades autoinmunes y sus títulos bajan en cuanto se resuelve el proceso infeccioso que les dio origen (92). El tercer grupo es el de los ANA autoinmunes, los cuales son secundarios a la pérdida de la tolerancia autoinmune y tienen un origen multifactorial (94). Actualmente, la técnica más utilizada para la detección de los ANA es la inmunofluorescencia indirecta (IFI), la cual fue desarrollada en 1950 por Coons et al. (93), y empleaba como sustratos cortes de hígado o riñón de ratón (95). En los últimos tiempos, la detección de los ANA se hace empleando como sustratos las líneas celulares HEp-2 y HeLa, siendo la primera por su facilidad de crecimiento la más utilizada. La detección de ANA mediante IFI en líneas celulares se considera la prueba inicial de laboratorio que apoya al diagnóstico de las enfermedades autoinmunes debido a su alta sensibilidad. Sin embargo, dada su relativa baja especificidad, es necesario emplear técnicas más sensibles y específicas para aumentar la sensibilidad y especificidad de los ANA para el diagnóstico (96).
Los patrones de fluorescencia de los ANA más frecuentes en el LES son el patrón homogéneo, periférico, y moteado, entre otros; la diferenciación y titulación de estos son de ayuda diagnóstica. El patrón homogéneo o difuso es un patrón producido por anticuerpos dirigidos contra las histonas. El patrón periférico es producido por anti DNA cuyos antígenos pueden ser a) Anti-DNA nativo, relacionado con la estructura doble hélice del DNA, considerados los más específicos del LES; b) Anti-DNA bicatenario que se dirige contra el esqueleto del DNA c) DNA monocatenario, desnaturalizado, que reaccionan contra las bases de los ácidos nucleicos. El patrón moteado o granular es un patrón de los anticuerpos contra la ribonucleoproteína. Hay diferentes variantes: a) patrón granular grueso (anti-RNP y anti-Sm) b) patrón granular fino (anti-Ro, anti-La) (97-99).
Los anticuerpos anti-DNA son marcadores importantes para el diagnóstico y el seguimiento de la actividad de la enfermedad, de hecho es el único anticuerpo que se puede utilizar para controlar la actividad de la misma. Niveles altos de anti-DNA, asociados a menudo a hipocomplementemia, se correlacionan con la actividad del LES así como con la presencia de nefritis lúpica (100). El antígeno Smith (Sm) es altamente específico para el LES pero es hallado solo en 25% de los pacientes con LES. El antígeno U1RNP es también encontrado en los pacientes con LES así como en la esclerosis sistémica o la enfermedad mixta del tejido conectivo. Los antígenos nucleares SSA (Ro) y SSB (La) son hallados en los pacientes con LES así como en los pacientes con síndrome de Sjögren. La presencia de anti-Ro/anti-La está asociado Lupus eritematoso subcutáneo pero sin actividad renal severa; así como también con LES neonatal, en el cual hay traspaso placentario de estos anticuerpos causando rash fotosensible, bloqueo cardiaco congénito o ambos (101,102) .
El sistema del complemento juega un papel más que importante en el LES. El rol del complemento es complejo ya que el mismo puede tanto prevenir como tener una participación en la exacerbación de la enfermedad. Los anticuerpos dirigidos contra el DNA y las histonas son comúnmente los causantes de la mayoría de las manifestaciones clínicas. Estos anticuerpos inducen la activación del complemento pero también la reacción inmunomediada del receptor Fc gamma (FcγR). De mayor relevancia, estos anticuerpos forman inmunocomplejos que se depositan en el glomérulo renal. Estos inmunocomplejos activan la vía clásica del complemento y causan daño tisular, lo cual lleva a nefritis lúpica (35,103,104) . La cascada del complemento es parte del sistema inmune innato. La deficiencia genética del iniciador de la cascada C1q predispone fuertemente al LES. Las deficiencias o mutaciones en otros componentes de la vía clásica, como C1r, C1s, C4 y C2 también incrementan el riesgo aunque en menor proporción en comparación con la deficiencia de C1q (105, 106).
Criterios de Clasificación
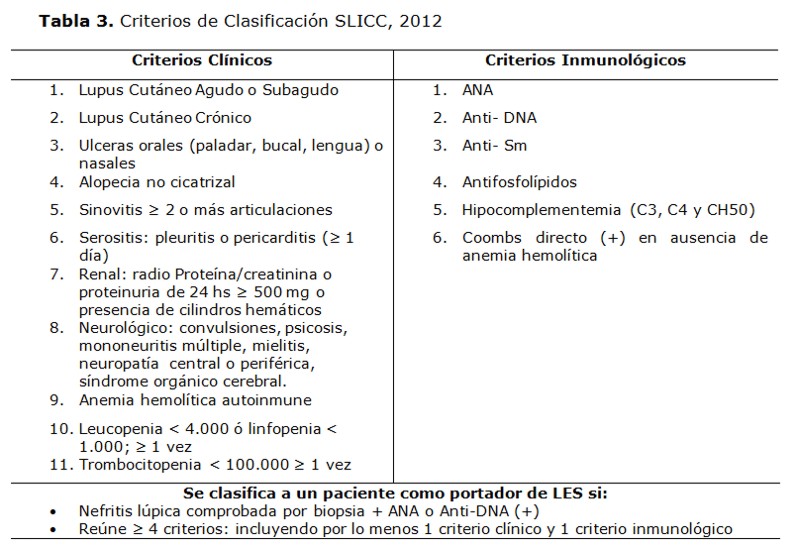
El diagnóstico del LES se basa en criterios definidos, que han experimentado una gran evolución a lo largo de los años. Los criterios de clasificación para el LES actualmente utilizados son los del Systemic Lupus International Collaborating Clinics Group (SLICC). Este es un grupo internacional de investigadores dedicado a la investigación clínica del LES, que analizó varias de las principales preocupaciones acerca de los criterios clínicos utilizados previamente, que incluían una posible duplicación de términos relacionados con el lupus cutáneo (como erupción malar y fotosensibilidad) y la falta de inclusión de muchas otras manifestaciones cutáneas, la omisión de muchas manifestaciones neurológicas y la necesidad de utilizar nuevos estándares en la cuantificación de proteinuría en la orina (36). Las preocupaciones acerca de los criterios inmunológicos incluían la omisión de los niveles de complemento bajo y la necesidad de incluir nueva información relativa a los anticuerpos antifosfolipídicos (36, 105) . Finalmente, se pensó que la nefritis confirmada por biopsia compatible con LES en presencia de autoanticuerpos para esta enfermedad, era tan indiscutiblemente representante de la enfermedad que debía ser considerada suficiente como criterio clínico (36,105) . Así pues los criterios SLICC tienen una sensibilidad superior a los criterios previos (SLICC 94% vs ACR 86%), sin embargo en cuanto a la especificidad es semejante; ACR 93% y SLICC 92%. Al utilizar los criterios de clasificación SLICC se consigue un menor número de errores con respecto a la clasificación de los pacientes con LES; por lo tanto los criterios SLICC que se observan en la Tabla 3 (106) representan mejor el compromiso orgánico e inmunológico del LES permitiendo también un diagnóstico más temprano.
CONCLUSIÓN
El lupus eritematoso sistémico es una enfermedad inflamatoria multisistémica, que se asocia a múltiples manifestaciones clínicas. La piel o las membranas mucosas, las articulaciones, el cerebro, el corazón, el riñón, el pulmón y ocasionalmente el tracto gastrointestinal pueden estar afectados. La inflamación de varios órganos o tejidos es secundaria a las alteraciones inmunológicas tanto del sistema inmune innato como del adaptativo. Las pruebas de laboratorio son de gran valor cuando se evalúa a un paciente con sospecha de enfermedad autoinmune, ya que los resultados pueden confirmar el diagnóstico, estimar la severidad de la enfermedad, evaluar el pronóstico y son de gran valor para el seguimiento de la actividad del LES. El conocimiento adecuado de la manifestaciones clínicas y laboratoriales en pacientes con LES nos permitirá realizar un mejor diagnóstico en los inicios de la enfermedad.
REFERENCIAS BIBLIOGRÁFICA
1. Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME. The geoepidemiology of systemic lupus erythematosus. Autoimmunity Reviews. 2010;9(5):A277-A87. [ Links ]
2. Fernández-Nebro A, Marsal S, Chatham W, Rahman A. Systemic Lupus Erythematosus: Genomics, Mechanisms, and Therapies. Clinical and Developmental Immunology. 2012;2012:1-2. [ Links ]
3. Gonzalez Martinez-Pedrayo AL. Bases genéticas de las enfermedades reumáticas. In: Blanco FJ, Cañete JD, Pablos JL, editors. Técnicas de investigación básica en reumatología. Madrid: Editorial médica Panamericana; 2007. p. 3-10. [ Links ]
4. Alarcón Riquelme ME. Genética del lupus eritematoso generalizado. ¿Qué se sabe y a dónde se va? Reumatología Clínica. 2010;6(1):1-2. [ Links ]
5. Fraser PA, Ding WZ, Mohseni M, Treadwell EL, Dooley MA, St Clair EW, et al. Glutathione S-transferase M null homozygosity and risk of systemic lupus erythematosus associated with sun exposure: a possible gene-environment interaction for autoimmunity. J Rheumatol. 2003;30(2):276-82. [ Links ]
6. Schoenfeld SR, Kasturi S, Costenbader KH. The epidemiology of atherosclerotic cardiovascular disease among patients with SLE: a systematic review. Semin Arthritis Rheum. 2013;43(1):77-95. [ Links ]
7. Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976;60(2):221-5 [ Links ]
8. Pons-Estel BA, Catoggio LJ, Cardiel MH, Soriano ER, Gentiletti S, Villa AR, et al. The GLADEL multinational Latin American prospective inception cohort of 1,214 patients with systemic lupus erythematosus: ethnic and disease heterogeneity among "Hispanics". Medicine (Baltimore). 2004;83(1):1-17. [ Links ]
9. Devins GM, Edworthy SM. Illness intrusiveness explains race-related quality-of-life differences among women with systemic lupus erythematosus. Lupus. 2000;9(7):534-41. [ Links ]
10. Ginzler EM, Diamond HS, Weiner M, Schlesinger M, Fries JF, Wasner C, et al. A multicenter study of outcome in systemic lupus erythematosus. I. Entry variables as predictors of prognosis. Arthritis Rheum. 1982;25(6):601-11. [ Links ]
11. Halberg P, Alsbjorn B, Balslev JT, Lorenzen I, Gerstoft J, Ullman S, et al. Systemic lupus erythematosus. Follow-up study of 148 patients. II: Predictive factors of importance for course and outcome. Clin Rheumatol. 1987;6(1):22-6. [ Links ]
12. Karlson EW, Daltroy LH, Lew RA, Wright EA, Partridge AJ, Fossel AH, et al. The relationship of socioeconomic status, race, and modifiable risk factors to outcomes in patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40(1):47-56. [ Links ]
13. McCarty DJ, Manzi S, Medsger TA, Jr., Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum. 1995;38(9):1260-70. [ Links ]
14. Molokhia M, Hoggart C, Patrick AL, Shriver M, Parra E, Ye J, et al. Relation of risk of systemic lupus erythematosus to west African admixture in a Caribbean population. Hum Genet. 2003;112(3):310-8. [ Links ]
15. Alarcon GS, Roseman J, Bartolucci AA, Friedman AW, Moulds JM, Goel N, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum. 1998;41(7):1173-80. [ Links ]
16. Peralta-Ramirez MI, Jimenez-Alonso J, Godoy-Garcia JF, Perez-Garcia M. The effects of daily stress and stressful life events on the clinical symptomatology of patients with lupus erythematosus. Psychosom Med. 2004;66(5):788-94. [ Links ]
17. Reveille JD, Moulds JM, Ahn C, Friedman AW, Baethge B, Roseman J, et al. Systemic lupus erythematosus in three ethnic groups: I. The effects of HLA class II, C4, and CR1 alleles, socioeconomic factors, and ethnicity at disease onset. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum. 1998;41(7):1161-72. [ Links ]
18. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54(8):2550-7. [ Links ]
19. Black L, Katz S. Respiratory Disease: Task Force Report on Problems,Research Approaches, Needs. DHEW report NIH77-1248. Arlington, VA: National Heart and Lung Institute; 1977. DHEW report NIH77-1248. [ Links ]
20. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol. (Review). 2015;advance online publication. [ Links ]
21. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384(9957):1878-88. [ Links ]
22. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus-an update. Current Opinion in Immunology. 2012;24(6):651-7. [ Links ]
23. Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, et al. SLE--a disease of clearance deficiency? Rheumatology (Oxford). 2005;44(9):1101-7. Epub 2005 May 31. [ Links ]
24. Sullivan KE. Genetics of systemic lupus erythematosus. Clinical implications. Rheum Dis Clin North Am. 2000;26(2):229-56, v-vi. [ Links ]
25. Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res. 2002;4(Suppl 3):S279-93. [ Links ]
26. Niu Z, Zhang P, Tong Y. Value of HLA-DR genotype in systemic lupus erythematosus and lupus nephritis: a meta-analysis. Int J Rheum Dis. 2015;18(1):17-28. [ Links ]
27. Lee YH, Bae SC. Association between the functional ITGAM rs1143679 G/A polymorphism and systemic lupus erythematosus/lupus nephritis or rheumatoid arthritis: an update meta-analysis. Rheumatol Int. 2015;35(5):815-23. [ Links ]
28. Ostanek L, Ostanek-Panka M, Bobrowska-Snarska D, Binczak-Kuleta A, Fischer K, Kaczmarczyk M, et al. PTPN22 1858C>T gene polymorphism in patients with SLE: association with serological and clinical results. Mol Biol Rep. 2014;41(9):6195-200. [ Links ]
29. Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204-10. [ Links ]
30. Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977-86. [ Links ]
31. Klemperer P, Pollack A, Baehr G. Diffuse collagen disease. Acute disseminated lupus erythematosus and diffuse scleroderma. JAMA. 1942:331-2. [ Links ]
32. Cutolo M, Sulli A, Straub RH. Estrogen metabolism and autoimmunity. Autoimmun Rev. 2012;11(6-7):A460-4. [ Links ]
33. Mason LJ, Isenberg D. The pathogenesis of systemic lupus erythematosus. In: Davidson AM, Cameron JS, Grunfeld JP, editors. Oxford textbook of clinical nephrology. Oxford, England: Oxford University Press; 2005. p. 809-29. [ Links ]
34. Rubin R. Drug induced lupus. In: Wallace DJ, Hahn BH, editors. Dubois’ lupus erythematosus. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2002. p. 885-916. [ Links ]
35. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271-7. [ Links ]
36. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. [ Links ]
37. Kuwana M, Kaburaki J, Okazaki Y, Miyazaki H, Ikeda Y. Two types of autoantibody-mediated thrombocytopenia in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2006;45(7):851-4. [ Links ]
38. Musio F, Bohen EM, Yuan CM, Welch PG. Review of thrombotic thrombocytopenic purpura in the setting of systemic lupus erythematosus. Semin Arthritis Rheum. 1998;28(1):1-19. [ Links ]
39. Villa Blanco I, Calvo Alén J. Lupus eritematoso sistémico. In: Alperi Lopez M, Balsa Criado A, Blanco A, Hernandez Cruz B, Medina Luezas J, Muñoz-Fernández S, et al., editors. Manual SER de enfermedades reumáticas. 6 ed. Madrid: Elsevier; 2014. p. 335-62. [ Links ]
40. Krupp LB, LaRocca NG, Muir J, Steinberg AD. A study of fatigue in systemic lupus erythematosus. J Rheumatol. 1990;17(11):1450-2. [ Links ]
41. Kuhn A, Landmann A. The classification and diagnosis of cutaneous lupus erythematosus. J Autoimmun. 2014;48-49:14-9. [ Links ]
42. Provost TT. Nonspecific cutaneous manifestations of systemic lupus erythematosus. In: Khun A, Lehmann P, Ruzicka T, editors. Cutaneous lupus erythematosus: Springer; 2004. p. 93-106. [ Links ]
43. Werth VP. Clinical manifestations of cutaneous lupus erythematosus. Autoimmunity Reviews. 2005;4(5):296-302. [ Links ]
44. Kuhn A, Lehmann P, Ruzicka T. Clinical manifestations of cutaneous lupus erythematosus. In: Kuhn A, Lehmann P, Ruzicka T, editors. Cutaneous lupus erythematosus: Springer; 2004. p. 59-92. [ Links ]
45. Horne NS, Narayan AR, Young RM, Frieri M. Toxic epidermal necrolysis in systemic lupus erythematosus. Autoimmun Rev. 2006;5(2):160-4. [ Links ]
46. Gronhagen CM, Fored CM, Linder M, Granath F, Nyberg F. Subacute cutaneous lupus erythematosus and its association with drugs: a population-based matched case-control study of 234 patients in Sweden. Br J Dermatol. 2012;167(2):296-305. [ Links ]
47. Tebbe B, Mansmann U, Wollina U, Auer-Grumbach P, Licht-Mbalyohere A, Arensmeier M, et al. Markers in cutaneous lupus erythematosus indicating systemic involvement. A multicenter study on 296 patients. Acta Derm Venereol. 1997;77(4):305-8. [ Links ]
48. Viguier M, Pinquier L, Cavelier-Balloy B, de la Salmoniere P, Cordoliani F, Flageul B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. A study of the relationship to lupus erythematosus. Medicine (Baltimore). 2001;80(3):180-8. [ Links ]
49. Kuhn A, Bein D, Bonsmann G. The 100th anniversary of lupus erythematosus tumidus. Autoimmun Rev. 2009;8(6):441-8. [ Links ]
50. Kuhn A, Richter-Hintz D, Oslislo C, Ruzicka T, Megahed M, Lehmann P. Lupus erythematosus tumidus--a neglected subset of cutaneous Lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136(8):1033-41. [ Links ]
51. Kuhn A, Ruland V, Bonsmann G. Photosensitivity, phototesting, and photoprotection in cutaneous lupus erythematosus. Lupus. 2010;19(9):1036-46. [ Links ]
52. Pipili C, Sfritzeri A, Cholongitas E. Deforming arthropathy in systemic lupus erythematosus. Eur J Intern Med. 2008;19(7):482-7. [ Links ]
53. Giannouli S, Voulgarelis M, Ziakas PD, Tzioufas AG. Anaemia in systemic lupus erythematosus: from pathophysiology to clinical assessment. Ann Rheum Dis. 2006;65(2):144-8. [ Links ]
54. Voulgarelis M, Kokori SI, Ioannidis JP, Tzioufas AG, Kyriaki D, Moutsopoulos HM. Anaemia in systemic lupus erythematosus: aetiological profile and the role of erythropoietin. Ann Rheum Dis. 2000;59(3):217-22. [ Links ]
55. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599-608. [ Links ]
56. Brey RL, Holliday SL, Saklad AR, Navarrete MG, Hermosillo-Romo D, Stallworth CL, et al. Neuropsychiatric syndromes in lupus: prevalence using standardized definitions. Neurology. 2002;58(8):1214-20. [ Links ]
57. Carbotte RM, Denburg SD, Denburg JA. Prevalence of cognitive impairment in systemic lupus erythematosus. J Nerv Ment Dis. 1986;174(6):357-64. [ Links ]
58. Appenzeller S, Cendes F, Costallat LT. Cognitive impairment and employment status in systemic lupus erythematosus: a prospective longitudinal study. Arthritis Rheum. 2009;61(5):680-7. [ Links ]
59. Devinsky O, Petito CK, Alonso DR. Clinical and neuropathological findings in systemic lupus erythematosus: the role of vasculitis, heart emboli, and thrombotic thrombocytopenic purpura. Ann Neurol. 1988;23(4):380-4. [ Links ]
60. Ellis SG, Verity MA. Central nervous system involvement in systemic lupus erythematosus: a review of neuropathologic findings in 57 cases, 1955--1977. Semin Arthritis Rheum. 1979;8(3):212-21. [ Links ]
61. Trysberg E, Nylen K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48(10):2881-7. [ Links ]
62. Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA, 3rd, Koffler D. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983;74(5):837-44. [ Links ]
63. Abreu MR, Jakosky A, Folgerini M, Brenol JC, Xavier RM, Kapczinsky F. Neuropsychiatric systemic lupus erythematosus: correlation of brain MR imaging, CT, and SPECT. Clin Imaging. 2005;29(3):215-21. [ Links ]
64. Ainiala H, Dastidar P, Loukkola J, Lehtimaki T, Korpela M, Peltola J, et al. Cerebral MRI abnormalities and their association with neuropsychiatric manifestations in SLE: a population-based study. Scand J Rheumatol. 2005;34(5):376-82. [ Links ]
65. Hanly JG, Urowitz MB, Siannis F, Farewell V, Gordon C, Bae SC, et al. Autoantibodies and neuropsychiatric events at the time of systemic lupus erythematosus diagnosis: results from an international inception cohort study. Arthritis Rheum. 2008;58(3):843-53. [ Links ]
66. Isshi K, Hirohata S. Association of anti-ribosomal P protein antibodies with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 1996;39(9):1483-90. [ Links ]
67. Yoshio T, Hirata D, Onda K, Nara H, Minota S. Antiribosomal P protein antibodies in cerebrospinal fluid are associated with neuropsychiatric systemic lupus erythematosus. J Rheumatol. 2005;32(1):34-9. [ Links ]
68. Hirohata S, Arinuma Y, Takayama M, Yoshio T. Association of cerebrospinal fluid anti-ribosomal p protein antibodies with diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res Ther. 2007;9(3):R44. [ Links ]
69. Petri M, Kasitanon N, Lee SS, Link K, Magder L, Bae SC, et al. Systemic lupus international collaborating clinics renal activity/response exercise: development of a renal activity score and renal response index. Arthritis Rheum. 2008;58(6):1784-8. [ Links ]
70. Ordi-Ros J, Torres MT, Segarra A, Vilardell M. Nefropatía lúpica. In: Cervera R, Jimenez-Alonso J, editors. Avances en lupus eritematoso sistémico. Barcelona: Marge Medica Books; 2011. p. 97-122. [ Links ]
71. Christopher-Stine L, Siedner M, Lin J, Haas M, Parekh H, Petri M, et al. Renal biopsy in lupus patients with low levels of proteinuria. J Rheumatol. 2007;34(2):332-5. [ Links ]
72. Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65(2):521-30. [ Links ]
73. Fries JF, Powers R, Kempson RL. Late-stage lupus nephropathy. J Rheumatol. 1974;1(2):166-75. [ Links ]
74. Sultan SM, Ioannou Y, Isenberg DA. A review of gastrointestinal manifestations of systemic lupus erythematosus. Rheumatology (Oxford). 1999;38(10):917-32. [ Links ]
75. Kwok SK, Seo SH, Ju JH, Park KS, Yoon CH, Kim WU, et al. Lupus enteritis: clinical characteristics, risk factor for relapse and association with anti-endothelial cell antibody. Lupus. 2007;16(10):803-9. [ Links ]
76. Breuer GS, Baer A, Dahan D, Nesher G. Lupus-associated pancreatitis. Autoimmun Rev. 2006;5(5):314-8. [ Links ]
77. Runyon BA, LaBrecque DR, Anuras S. The spectrum of liver disease in systemic lupus erythematosus. Report of 33 histologically-proved cases and review of the literature. Am J Med. 1980;69(2):187-94. [ Links ]
78. Tse KC, Yung S, Tang C, Yip TP, Chan TM. Management of hepatitis B reactivation in patients with lupus nephritis. Rheumatol Int. 2009;29(11):1273-7. [ Links ]
79. Kao AH, Manzi S. How to manage patients with cardiopulmonary disease? Best Pract Res Clin Rheumatol. 2002;16(2):211-27. [ Links ]
80. Memet B, Ginzler EM. Pulmonary manifestations of systemic lupus erythematosus. Semin Respir Crit Care Med. 2007;28(4):441-50. [ Links ]
81. Pan TL, Thumboo J, Boey ML. Primary and secondary pulmonary hypertension in systemic lupus erythematosus. Lupus. 2000;9(5):338-42. [ Links ]
82. Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus. 2005;14(9):683-6. [ Links ]
83. Wijetunga M, Rockson S. Myocarditis in systemic lupus erythematosus. Am J Med. 2002;113(5):419-23. [ Links ]
84. Makaryus JN, Catanzaro JN, Goldberg S, Makaryus AN. Rapid progression of atrioventricular nodal blockade in a patient with systemic lupus erythematosus. Am J Emerg Med. 2008;26(8):967.e5-7. [ Links ]
85. Moder KG, Miller TD, Tazelaar HD. Cardiac involvement in systemic lupus erythematosus. Mayo Clin Proc. 1999;74(3):275-84. [ Links ]
86. Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA, Jr., Jansen-McWilliams L, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997;145(5):408-15. [ Links ]
87. Magder LS, Petri M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am J Epidemiol. 2012;176(8):708-19. [ Links ]
88. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721-31. [ Links ]
89. Castro C, Gourley M. Diagnostic testing and interpretation of tests for autoimmunity. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S238-47. [ Links ]
90. Tan EM. Autoantibodies to nuclear antigens (ANA): their immunobiology and medicine. Adv Immunol. 1982;33:167-240. [ Links ]
91. Conrad K, Schöbler W, Hiepe F, Fritzler MJ. Autoantigens, autoantibodies, autoimmunity. Autoantibodies in systemic autoimmune diseases. A diagnostic reference: Pabst Science Publisher; 2002. [ Links ]
92. Shoenfeld Y, Meroni P, Gershwin M. Autoantibodies. 2n ed. Amsterdam, Boston: Elsevier; 2007. [ Links ]
93. Coons AH, Kaplan MH. Localization of antigen in tissue cells; improvements in a method for the detection of antigen by means of fluorescent antibody. J Exp Med. 1950;91(1):1-13. [ Links ]
94. Cabiedes J, Núñez-Álvarez CA. Anticuerpos antinucleares. Reumatol Clín. 2010;6(4):224-30. [ Links ]
95. Damoiseaux JG, Tervaert JW. From ANA to ENA: how to proceed? Autoimmun Rev. 2006;5(1):10-7. [ Links ]
96. Morehead K. Evaluation of the patient. Laboratory Assessment. In: Klippel JH, Stone JH, White PH, editors. Primer on the rheumatic diseases. 13th ed. New York: Springer Science & Business Media; 2008. p. 15-21. [ Links ]
97. Reeves WH, Satoh M, Richards HB. Origins of antinuclear antibodies. In: Lahita RG, Tsokos G, Buyon J, Koike T, editors. Systemic lupus erythematosus. San Diego: Academic Press; 2004. p. 401-31. [ Links ]
98. Gullstrand B, Lefort MH, Tyden H, Jonsen A, Lood C, Johansson A, et al. Combination of autoantibodies against different histone proteins influences complement-dependent phagocytosis of necrotic cell material by polymorphonuclear leukocytes in systemic lupus erythematosus. J Rheumatol. 2012;39(8):1619-27. [ Links ]
99. Jovanovic V, Dai X, Lim YT, Kemeny DM, MacAry PA. Fc gamma receptor biology and systemic lupus erythematosus. Int J Rheum Dis. 2009;12(4):293-8. [ Links ]
100. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785-97. [ Links ]
101. Julkunen H, Ekblom-Kullberg S, Miettinen A. Nonrenal and renal activity of systemic lupus erythematosus: a comparison of two anti-C1q and five anti-dsDNA assays and complement C3 and C4. Rheumatol Int. 2012;32(8):2445-51. [ Links ]
102. Li J, An L, Zhang Z. Usefulness of complement activation products in Chinese patients with systemic lupus erythematosus. Clin Exp Rheumatol. 2014;32(1):48-53. [ Links ]
103. Batal I, Liang K, Bastacky S, Kiss LP, McHale T, Wilson NL, et al. Prospective assessment of C4d deposits on circulating cells and renal tissues in lupus nephritis: a pilot study. Lupus. 2012;21(1):13-26. [ Links ]
104. McPherson RA, Pincus MR. Henry's clinical diagnosis and management by laboratory methods. 21 ed. Philadelphia: WB Saunders; 2007. [ Links ]
105. Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39(3):363-9. [ Links ]
106. Petri M, Magder L. Classification criteria for systemic lupus erythematosus: a review. Lupus. 2004;13(11):829-37. [ Links ]
Fecha de recepción: noviembre 2015. Fecha de aceptación: febrero 2016
Autor correspondiente: *Osmar Antonio Centurión. Departamento de Medicina Interna. Hospital de Clínicas. Universidad Nacional de Asunción
E-mail: osmarcenturion@hotmail.com

