










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
On-line version ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud vol.13 no.1 Asunción Apr. 2015
https://doi.org/10.18004/Mem.iics/1812-9528/2015.013(01)83-087
REPORTE DE CASO
Tetralogía de Fallot asociada a duplicación distal del brazo largo del cromosoma 11
Tetralogy of Fallot associated with distal duplication of the long arm of chromosome 11
*Torres E, Rodríguez S, Monjagata N
Laboratorio de Genética. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción. Asunción, Paraguay
RESUMEN
La tetralogía de Fallot es una cardiopatía frecuente y puede representar hasta el 11 al 13% de todas las cardiopatías congénitas clínicas, se presenta en 1 de cada 8.500 nacidos vivos. En la mayoría de los casos, se asocia a una microdeleción del cromosoma 22 y con menor frecuencia al síndrome de Down. El síndrome de la dup 11q es una anomalía cromosómica causada por la duplicación de la porción distal del extremo del brazo largo del cromosoma 11, ocasionando una trisomía parcial del mismo, producto de un desbalance cromosómico, con disfunción de los genes involucrados en este material genético adicional que ocasiona anormalidades tanto físicas como mentales en un recién nacido. Se presenta el caso de un niño de 3 meses de vida que es derivado a la consulta genética por fenotipo sindromático, Tetralogía de Fallot y retraso del crecimiento. El estudio citogenético se realizó en sangre periférica, los cromosomas fueron procesados con técnicas de tinción convencional, bandas de alta resolución y centroméricas, observándose una duplicación 11q. Cariotipo: 46, XY, dup11 (q23àqter). Se enfatiza la importancia del estudio cromosómico en recién nacidos con malformaciones congénitas mayores para el diagnóstico de certeza y posterior asesoramiento genético a los progenitores.
Palabras clave: duplicación, trisomía, cromosoma 11q.
ABSTRACT
Tetralogy of Fallot is a common heart disease and may represent up to 11 to 13% of all clinical congenital cardiopathies, it occurs in about 1 out of every 8,500 live births. In most cases is associated with a microdeletion of chromosome 22 and less frequently with Down syndrome. Syndrome Dup11q is a chromosomal abnormality caused by duplication of the distal end of the long arm of chromosome 11 resulting in a partial trisomy, product of a chromosomal imbalance, with dysfunction of the genes involved in this additional genetic material causing both physical and mental abnormalities in a newborn. This is the case of a 3-month boy who was referred to genetic consultation due to syndromic phenotype, Tetralogy of Fallot and growth retardation. The cytogenetic study was performed in peripheral blood. Chromosomes were processed with conventional staining techniques, centromeric and high-resolution bands, showing 11q duplication. Karyotype: 46, XY, dup11 (q23àqter). We emphasize the importance of chromosomal studies in infants with major congenital malformations for a subsequent accurate diagnosis and genetic counseling to parents.
Keywords: duplication, trisomy, chromosome 11q.
INTRODUCCIÓN
Las malformaciones congénitas y las enfermedades genéticas son una causa importante de morbilidad y mortalidad infantil, sobre todo en la actualidad y en países del primer mundo, en donde las infecciones pasaron a segundo plano. En nuestro país, las malformaciones congénitas constituyen la cuarta causa de mortalidad infantil (1). Las anomalías cromosómicas están asociadas a malformaciones congénitas mayores, principalmente a las del aparato cardiovascular. Una de estas malformaciones cardíacas, la Tetralogía de Fallot, consiste en un defecto en el que se describen cuatro anomalías, a) estrechez de la válvula pulmonar, b) engrosamiento de la pared ventricular derecha, c) desplazamiento de la aorta sobre el defecto del tabique ventricular y d) apertura del defecto del tabique ventricular entre los ventrículos izquierdo y derecho. Esto acarrea como consecuencia el bombeo de sangre insuficientemente oxigenada al cuerpo y se clasifica como un defecto cardiaco cianótico, porque los pacientes presentan una coloración azulada o púrpura de la piel y falta de aliento debido a los niveles bajos de oxígeno en la sangre (2).
El término duplicación en citogenética implica que parte de un cromosoma está repetido o presenta dos copias, teniendo como resultado material genético adicional, aún cuando el total de cromosomas esté aparentemente dentro de lo normal. El síndrome de la duplicación 11q es una anomalía cromosómica causada por la repetición de la porción distal del brazo largo del cromosoma 11, ocasionando un desbalance cromosómico con disfunción de los genes involucrados en este material adicional, provocando anormalidades tanto físicas como mentales y su origen puede ser al azar o estar asociado a reordenamientos cromosómicos en los progenitores (3,4).
Se describe el caso de un niño con diagnóstico clínico de Tetralogía de Fallot, a quien se le realizó el estudio citogenético, resultando portador de una duplicación distal del brazo largo del cromosoma 11.
CASO CLINICO
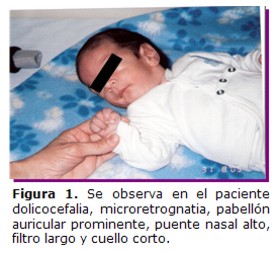
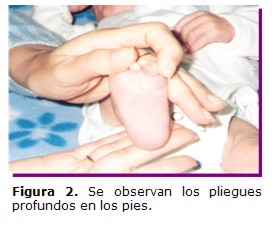
Paciente de sexo masculino, de 3 meses de vida, madre y padre de 30 años de edad, no consanguíneos, con antecedente de un aborto espontaneo de causa desconocida, no hay antecedentes patológicos del embarazo actual, ni de ingestión de medicamentos. Si presentó placenta de inserción baja, con hemorragias leves durante el tercer y cuarto mes. Al examen físico el niño presentó el siguiente fenotipo: circunferencia cefálica 39,5 cm percentilo 25; talla 57 cm. percentilo 10; peso 3,5 Kg, dolicocefalia, puente nasal alto, orejas de implantación baja, microretrognatia, fontanela anterior amplia, pliegues palmares únicos en manos y profundos en pies (Figuras 1 y 2) y soplo cardíaco; por ecocardiografía se detectó la Tetralogía de Fallot.
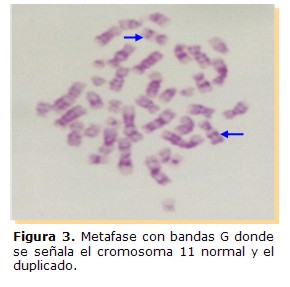
El cariotipo se realizó a través del cultivo de linfocitos de sangre periférica, los cromosomas fueron teñidos con Giemsa para el estudio convencional y la identificación cromosómica se llevó cabo con técnicas de Bandas C y de Alta Resolución. Esta última se realizó debido a que con esta técnica se pueden visualizar hasta 2.000 bandas, haciendo posible detectar anomalías de menor tamaño (5). Fueron analizadas treinta células del paciente con tinción convencional, encontrándose en todas ellas 46 cromosomas. Con la técnica de Alta Resolución se detectó en todas las metafases la presencia de una duplicación en la región distal del brazo largo de uno de los cromosomas del par 11, desde la región q,23.1 hasta la porción terminal (Figuras 3 y 4).
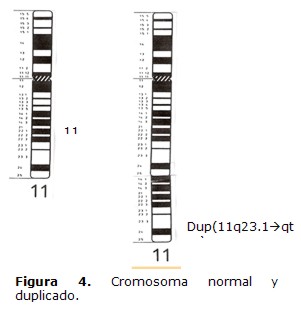
El cariotipo del niño fue 46, XY, dup11 (q23,1à qter) mientras que el de los padres fue normal, 46,XX para la madre y 46,XY para el padre.
DISCUSIÓN
Entre una de las anomalías cardíacas se describe la Tetralogía de Fallot (TOF), que es una cardiopatía frecuente y puede representar de un 11 a 13% de todas las cardiopatías congénitas clínicas, que se estima ocurre en 1 de cada 8.500 nacidos vivos. Junto con las transposición de grandes arterias es la cardiopatía cianótica más frecuente. En aproximadamente 16% de los casos se asocia a una microdelecion del cromosoma 22, pero también se asocia con menos frecuencia al síndrome de Down (6). Este defecto congénito puede ser causado por una mutación heterocigota en los cromosomas 20p12 (gen JAG1), 5q35.1 (gen NKX2-5), 8q23.1 (gen ZFPM2), 18,q11 (gen GATA6); 19p13 (gen GDF1). También se han descrito la presencia de la Tetralogía de Fallot en duplicaciones de los cromosomas 1q21 y 8q22, pero es una característica bien conocida del síndrome de microdeleción del cromosoma 22q11 (gen TBX1) y de la trisomía 21 (7). Johnson en 1997 llevó a cabo una evaluación citogenética a 159 casos con Tetralogía de Fallot, identificando la deleción 22q11 en el 14% de los pacientes que se realizaron la Hibridación in situfluorescente (FISH)(8). Rauch et al. en el 2.010 encontraron que la deleción 22q11.2 era la anomalía genética más común entre 230 pacientes con TOF, observándose en el 7,4% de los pacientes y la segunda anomalía más frecuente fue la trisomía 21, que se observó en el 5,2% de los pacientes (9).
En nuestro paciente, la Tetralogía de Fallot está asociada a la duplicación del cromosoma 11q, la cual es un rearreglo cromosómico que se origina por emparejamiento y recombinación asimétricos, en algunos casos a partir de translocaciones balanceadas en los padres, la mayoría de origen materno, y en otros casos, los menos son mutaciones de novo; como en este caso, pues el cariotipo de los padres ha resultado normal.
La escasez de casos y la inespecificidad de las manifestaciones clínicas en los pacientes portadores de la duplicación del cromosoma 11q, hacen difícil delinear un fenotipo clínicamente reconocible Se describe que alrededor del 85% tiene orejas de implantación baja, el 60% presenta paladar hendido, más del 50% presenta retraso del desarrollo mental y físico pre y post natal con rangos que van desde retraso mental leve a severo, hipertonía en el 50%, asimetría craneofacial y microcefalia en el 35% de los individuos afectados Pueden, además, presentar un fenotipo no muy constante, como hipertelorismo, pliegues epicanticos y fisuras palpebrales oblicuas, nariz chata y filtrumlargo, orificios o mamelones preauriculares generalmente bilaterales, labios finos y micrognatia y el cuello es generalmente corto. El retardo mental presente en estos pacientes varía de leve a severo (3, 4, 10-17). La mayoría de los pacientes con TOF no requiere tratamiento en el periodo neonatal y puede darse de alta al domicilio con revisiones cardiológicas frecuentes, el tratamiento depende del grado de severidad del cuadro, pero en general si existe una cianosis severa o progresiva y la presencia de crisis hipoxémicas se indica un tratamiento quirúrgico (6)
En relación a pacientes portadores de la duplicación distal del cromosoma 11, la literatura revisada reporta 6/16 que murieron en el primer año de vida, el de mayor edad murió a los 16 años, con una trisomía para 11q21àqter. La severidad de las características clínicas y el pronóstico son relativos, dependen de la extensión del segmento duplicado (10,14).
Con este trabajo se resalta el valor del estudio citogenético en recién nacidos portadores de malformaciones congénitas mayores, para el diagnóstico de certeza y posterior asesoramiento genético a los progenitores.
REFERENCIAS BIBLIOGRÁFICAS
1. Fonseca R, Mir R, Irala S, Navarro E, Ortigosa M, Céspedes E, et al. Conocimientos de la etiología y los factores de riesgos de los defectos congénitos en pediatría. Pediatr. (Asunción). 2008; 35(2): 95-100. [ Links ]
2. Gonzalez JA, Cadavid AM, Aguilera D, Cazzaniga M. Articulo de actualización para formación continuada tetralogía de Fallot. Rev. Col.Cardiol. 2008; 15(3):139.47. [ Links ]
3. Louise Buyse M, editor. Birth defects encyclopedia. USA: Blackwell Scientific; 1990. [ Links ]
4. Klaassens M, Scott D, Van Dooren M, Hochstenbach R, Eussen HJ, Cai WWW, et al. Congenital diaphragmatic hernia associated with duplication of 11q23-qter. Am J Med Genet A. 2006; 140(14):1580-6. [ Links ]
5. Verma R, Babu A. Human chromosome: Manual of basic techniques. New York: Pergamon Press; 1989. [ Links ]
6. Rodriguez M, Villagrá F. Capitulo 11: Protocolos diagnósticos y terapéuticos en cardiología pediátrica. En: Tetralogía de Fallot /Internet/. /citado 10 oct 2014/. Disponible en: http://telecardiologo.com/descargas/66993.pdf [ Links ]
7. Johns Hopkins University, Online Mendelian Inheritance in Man. Duplication 11. En: An online catalog of human genes and genetic disorders. Updated 16 setiembre 2013. [ Links ]
8. Johnson M, Hing A, Wood M, Watson M. Chromosome abnormalities in congenital heart disease. Am J Med Genet. 1997; 70:292-8. [ Links ]
9. Rauch R, Hofbeck M, Zweier C, Koch A, Zink S, Trautmann U, et al. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J Med Genet. 2010; 47:321-31. [ Links ]
10. Gorlin RJ, Cohen MM Jr, Hennekam RCM. Syndromes of the head and the neck. 6th ed. USA: Oxford University Press; 2010. [ Links ]
11. Jehee F, Bertola D, Yelavarthi K, Krepischi-Santos A, Kim C, Vianna-Morgante A, et al. An 11q11-q13.3 duplication, including FGF3 and FGF4 genes, in patient with syndromic multiple craniosynostoses. Am J Med Gent A. 2007; 43(16):1912-8. [ Links ]
12. Zarate Y, Kogan J, Schorry E, Smolarek T, Hopkin R. A new case of de novo11q duplication in a patient with normal development ant intelliencie and review of the literature. Am J Med Genet A. 2007; 143(3):265-70. [ Links ]
13. Partida-Pérez M, Domínguez M, Sánchez-Corona J, Castañeda-Cisneros G, García-González C, Lopez Cardona M, et al. Constitucional duplication 11q23 de novoinvolving the MLL gene. Genet Couns. 2006; 17(2):155-9. [ Links ]
14. Jones KL. SMITH´S recognizable patterns of human malformation. 6th ed. USA: Elsevier Saunders; 2006. [ Links ]
15. Shashidhar G, Lewandowski R, Digamber SB. Handbook and chromosomal syndromes. USA: John Wiley & Sons; 2003. [ Links ]
16. Gorlin R, Cohen M, Hennekam R. Chromosomal syndromes: Unusual variants. En: Síndromes of the Head and Neck. 4th ed. New York: Oxford University Press; 2001. p. 100. [ Links ]
17. Yelavarthi K, Zunich J. Familial interstitial duplication of 11q; partial trisomy. (11)(q13.5q21). Am J Med Genet A. 2004; 126(4):423-6. [ Links ]
*Autor Correspondiente: Dra. Elodia Torres, Laboratorio de Genética. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción (IICS-UNA). Asunción, Paraguay
Email: genética@iics.una.py
Fecha de recepción: noviembre 2013; Fecha de aceptación: diciembre 2014
