










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
On-line version ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud vol.5 no.1 Asunción June 2007
REPORTE DE CASOS
El Síndrome de Williams-Reporte de tres casos
Williams syndrome- Report of three cases
Herreros MBI,II*, Ascurra MII, Franco RI
IInstituto Nacional de Protección a Personas Excepcionales(INPRO)
IIInstituto de Investigaciones en Ciencias de la Salud (IICS).Asunción-Paraguay
RESUMEN
El síndrome de Williams, síndrome de Williams- Beuren, hipercalcemia idiopática o síndrome de estenosis aórtica supravalvular, es un desorden de etiología genética caracterizado por facies dismórfica con características típicas, retraso mental, deficiencia del crecimiento, anomalías del tejido conectivo, anomalías cardiovasculares, una personalidad típica y, a veces, hipercalcemia en la infancia. La frecuencia de este síndrome ha sido estimada en 1 en 10000 a 20000 nacimientos. La causa del mismo, es una deleción submicroscópica del cromosoma 7q11-13 que incluye el gen de la elastina (ELN) en 90 a 95% de los casos. Este es un síndrome de genes contiguos (diferentes genes implicados, producen diferentes síntomas) y se conocen comprometidos los genes: ELN (implicado en las anomalías del tejido conectivo), GTF21 (implicado en el retardo mental) y LIMK1 (implicado en el fenotipo del síndrome). La deleción es de novo en la mayoría de los casos, aunque se ha documentado trasmisión de padres a hijos. Se puede realizar el diagnostico prenatal por Fluorescente in situ hybridization(FISH). CASOS CLINICOS: Se describen los casos de dos niños de 1 año y 9 meses y 1 año y 10 meses, y una niña de 6 años y 8 meses, los tres con diferentes características clínicas. En este trabajo se resalta la importancia del fenotipo sobre las otras características del síndrome, que pueden o no estar presentes.
Palabras claves: Estenosis aórtica supravalvular, deleción, elastina.
ABSTRACT
Williams syndrome, also known as Williams-Beuren syndrome, Idiopathic hypercalcemia or supravalvular aortic stenosis syndrome, is a genetic disorder characterized by dysmorphic facies with typical features, mental retardation, growth deficiencies, anomalies of connective tissue, cardiovascular anomalies, a typical personality and sometimes hypercalcemia during childhood. The frequency of this syndrome has been estimated as 1 in 10,000 to 20,000 births. The cause is a sub-microscopic deletion of 7q11-13 chromosome that includes the elastine gen (ELN) in 90 to 95% of cases. This is a syndrome of adjacent genes (different genes involved, produce different symptoms)and the genes involved are: ELN (involved in the anomalies of connective tissue), GTF21 (involved in the mental retardation) andLIMK1 (involved in the syndrome phenotype). In most cases the deletion is de novo, though transmission from parents to children has been reported. The prenatal diagnosis can be made by fluorescent in situ hybridization (FISH). Here we report the cases of a one-year-nine-month-old boy, another one-year-ten-month-old boy and a six-year-eight-month-old girl, the three of them with different clinical characteristics. This work emphasizes the importance of phenotype above all the other characteristics of the syndrome that may or may not be present.
Keywords: Supravalvular aortic stenosis, deletion, elastine.
INTRODUCCION
El síndrome de Williams (SW), síndrome de Williams- Beuren, Hipercalcemia Idiopática o Síndrome de Estenosis Aórtica Supravalvular, es un desorden de etiología genética, caracterizado por; facies dismórfica con características típicas, retraso mental, deficiencia del crecimiento, anomalías del tejido conectivo, anomalías cardiovasculares, personalidad típica y, a veces, hipercalcemia en la infancia. Se presenta tanto en niños, como en niñas1-6. La frecuencia de este síndrome ha sido estimada en 1 en 10000 a 20000 nacimientos. La causa del mismo, es una deleción submicroscópica del cromosoma 7q11-13 que incluye el gen de la elastina (ELN) en 90 a 95% de los casos. El síndrome de Williams es un síndrome de genes contiguos (diferentes genes implicados, producen diferentes síntomas) y se conocen comprometidos los genes: ELN (implicado en las anomalías del tejido conectivo), GTF21 (implicado en el retardo mental) y LIMK1 (implicado en el fenotipo del síndrome)4-6,8. El diagnóstico se realiza por la clínica y a través de estudios de laboratorio específicos como, Fluorescente in situ hybridization (FISH) del cromosoma 7 (que da positivo en el 99% de los casos), o por estudio de biología molecular. Se puede realizar el diagnostico prenatal por FISH. Los diagnósticos diferenciales tenidos en cuenta son; la estenosis aórtica supravalvular aislada, el síndrome de Noonan, FG, Kabuki, síndrome de alcohol fetal y síndrome del X frágil4.
CARACTERISTICAS CLINICAS
El SW tiene un fenotipo facial muy característico, llamado “carita de duende”, que consiste en; hinchazón periorbitaria, nariz corta de punta bulbosa, filtrum largo, boca ancha, labios llenos y micrognatia leve. En la infancia tienen perfil plano y los niños mayores, cara fina y cuello largo. Los pacientes de ojos azules o verdes, presentan un patrón estrellado, “iris stellate”. También presentan retraso del crecimiento, con un índice de crecimiento 75% del normal1-4, 6,7. La mayoría de los niños con SW tienen anomalías cardiovasculares, siendo la más común la estenosis aórtica supravalvular, que generalmente es progresiva y requiere tratamiento quirúrgico. Otras anomalías cardiacas son: la estenosis de las arterias pulmonares periféricas, coartación de aorta, estenosis de arterias renales e hipertensión arterial4, 6,9. En algunos casos se ve hipercalcemia en la infancia (15%) y esta puede provocar, vómitos, irritabilidad, constipación y calambres musculares. En el 80% de los casos se observa hipotonía de tronco y en el 50%, hipertonía periférica. Los reflejos osteotendinosos profundos suelen estar aumentados. Los niños con SW pueden tener reflujo gastroesofágico, dificultad para alimentarse o cólicos prolongados1,4,6. Estos niños presentan un perfil cognitivo y de conducta, muy típico. El perfil cognitivo consiste en una fortaleza relativa en la memoria auditiva y el lenguaje, y extrema debilidad en el área visuoespacial y constructiva. El problema de conducta consiste en; déficit de atención, ansiedad, actitud extremadamente sociable y el 73% de los casos presenta hiperactividad. El 75% de los niños SW presenta retraso mental y retraso del lenguaje en la infancia, aunque luego esta es su área más fuerte1,4,6,10-13. El 85-95% de los casos los niños tienen hipersensibilidad a los sonidos4,14. La voz es ronca y pueden tener laxitud o limitación del movimiento articular, maloclusión dental, hernias y piel suave y sobrante. Pueden presentar anomalías oftalmológicas como estrabismo (50%), errores refractivos como hiperopia y anormalidades en la visión binocular. En el 50% de los casos se reporta otitis media recurrente. Sus dedos suelen ser cortas con uñas cortas y anchas. Pueden tener anomalías renales como nefrocalcinosis, riñón único y defectos de vías urinarias. En los adultos se pueden observar signos de disfunción cerebelar y escoliosis1,4,6.
CASOS CLINICOS
CASO 1
Niña de 6 años y 8 meses de edad, que consulta por problemas de aprendizaje y fenotipo sindromático. La niña es el producto del 2º embarazo de una madre de 40 años, 5º hija de un padre de 52 años. Ambos padres aparentemente sanos y no consanguíneos. Antecedentes familiares: una hermana de padre y madre (9 a) sana y tres hermanos de padre; un hermano (26 a), una hermana (20 a) sanos y un hermano (25 a) con problemas de aprendizaje. No hay antecedentes patológicos del embarazo actual, ni de ingestión de medicamentos. Nació a las 39 semanas, en un parto por cesárea con un peso de 2500 grs., perc: 25, no hay datos de talla y circunferencia cefálica al nacimiento. El desarrollo sicomotor fue normal.
ESTUDIOS
Ecocardiografía: morfología normal, extrasístoles. Ecografía abdominal y renal: normal. Ecografía pelviana: ovario izquierdo aumentado de tamaño y con múltiples imágenes quísticas. Calcemia: normal. EEG: lento, en forma difusa. Examen de ojo: normal. Carácter alegre y sociable, muy conversadora.
EXAMEN FISICO
CC: 49cm. perc: 25 Talla: 111,5cm. perc: 10 y Peso: 15kg. perc: <p3 Cara triangular, cachetes prominentes, hinchazón periorbitaria, puente nasal alto, punta nasal bulbosa, labios llenos, boca ancha, micrognatia, orejas prominentes y de implantación baja, cuello largo. (Figuras 1 y 2). Manos: pliegues palmares equivalentes y uñas cortas y anchas. (Figura 3). Genitales normales. Se ausculta un soplo cardiaco. Es muy sociable y conversadora. Aun no se realizaron estudios específicos para el S. de Williams por motivos económicos.
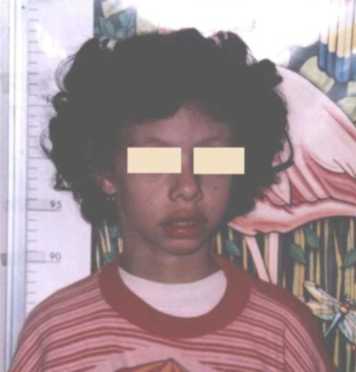
Figura 1. Nótese, labios llenos, punta nasal bulbosa, cuello largo.
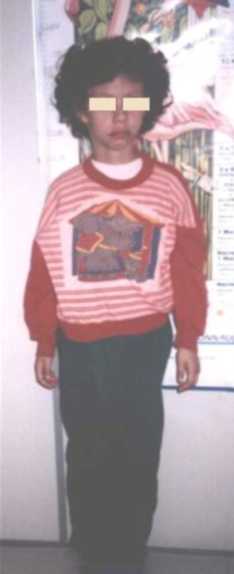
Figura 2. Talla baja
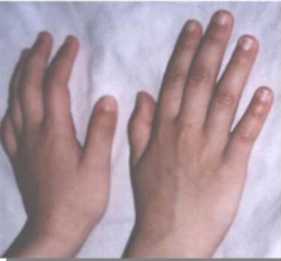
Figura 3. Uñas cortas y anchas.
CASO 2
Niño de 1 año y 9 meses de edad, que consulta por RDSM, estenosis aórtica supravalvular y reflujo gastroesofágico. El niño es el producto del 6º embarazo de una madre de 33 años, y un padre de 37 años. Ambos, aparentemente sanos y no consanguíneos. Antecedentes familiares: la madre tuvo tres abortos espontáneos, tiene una hermana (12 a) y un hermano (8 a) sanos. Antecedentes patológicos del embarazo actual; amenaza de aborto espontáneo desde el primer mes (muchas contracciones). Nació en un parto normal a las 38 semanas, con un peso de 2430 grs. perc: 3 talla: 46 cm., perc: 3 y CC: 32 cm. perc:-2DS. Al nacimiento presentó llanto espontáneo, hipotonía y succión débil. El desarrollo sicomotor fue lento, adquiriendo sostén cefálico a los 11 meses, sedestación sin apoyo al 1 año y en el momento de la consulta estaba empezando a caminar y decía algunas palabras sueltas5-6.
ESTUDIOS
Ecocardiografía: estenosis aórtica supravalvular, coartación de aorta moderada, sospecha de doble arco aórtico. Examen de orina: cristales de oxalato de calcio. TAC de cráneo, normal. Cariotipo: normal. FISH para el cromosoma 7; deleción 7 q11-23. Carácter alegre y sociable.
EXAMEN FISICO
CC: 45, 5 cm. perc: -2DS, Talla: 74 cm., perc:<p3, Peso: 12 kg. perc: 3-10. Cachetes prominentes, hinchazón periorbitaria, epicantus, puente nasal chato, nariz corta, punta nasal bulbosa, labios llenos, boca ancha, micrognatia (Figura 4). Manos: pliegues palmares normales y uñas cortas y anchas. Se ausculta un soplo cardiaco. Genitales normales. Es de carácter alegre y sociable.
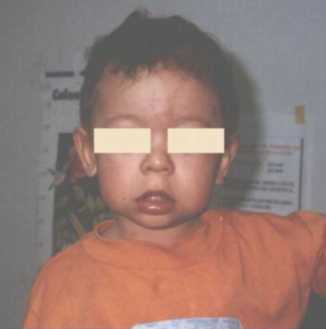
Figura 4.Labios llenos, punta nasal bulbosa, mejillas prominentes.
CASO 3
Niño de 1 año y 10 meses de edad, que consulta por RDSM y colpocefalia. El niño es el producto del 5º embarazo de una madre de 36 años, y el primer hijo de un padre de 44 años. Ambos, aparentemente sanos y no consanguíneos. Antecedentes familiares: la madre tiene un hijo de 16 años y un hijo de 8 años, ambos sanos, y dos abortos espontáneos, de otro padre. Su embarazo fue normal y no refiere haber ingerido medicamentos, ni alcohol. Nació en un parto normal a las 38 semanas, con un peso de 2500 grs. perc: 3, talla: 47 cm., perc: 3 y no hay datos de circunferencia cefálica al. Al nacimiento presentó llanto espontáneo, succión normal, sin complicaciones. El desarrollo sicomotor fue lento, adquiriendo sostén cefálico a los 5 meses, logró sedestación sin apoyo a los 8 meses y marcha al año y 3 meses. En el momento de la consulta aun no tenía lenguaje.
ESTUDIOS
EEG: en sueño profundo, con actividad paroxismal tipo PO, lenta generalizada. TAC de cráneo: normal. RMN de cerebro: colpocefalia y ensanchamiento de astas posteriores de ventrículos laterales. Ecocardiografía: Estenosis aórtica supravalvular, leve a moderada, sin signos de sobrecarga. Calcemia: normal.
EXAMEN FISICO
CC: 47 cm. perc: 25, talla: 84 cm., perc: 25 y peso: 12 kg., perc; 25-50. Se observa hinchazón periorbitaria, epicantus bilateral, hipoplasia medionasal, puente nasal chato, narinas antevertidas, filtrum liso, labios llenos, boca ancha y orejas prominentes con hélices dismórficos(Figura 5). Tiene uñas cortas y anchas, en las mano izquierda presenta un pliegue palmar normal y en la mano derecha un pliegue palmar único (Figura 6). Sus genitales son normales y tiene una hernia umbilical. Se ausculta un soplo cardiaco. Es de carácter alegre y muy sociable. Aun no se realizaron los estudios de diagnostico específicos para el SW, por motivos económicos.
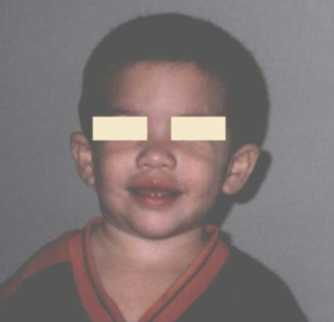
Figura 5. Véase hinchazón periorbitaria, labios llenos y mejillas prominentes.
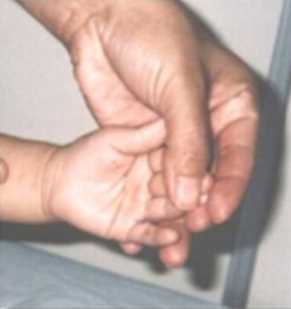
Figura 6. Pliegue palmar único.
CONCLUSION
En los tres casos, el diagnóstico clínico es de síndrome de Williams. En el caso 1, el diagnóstico se realizó por el fenotipo facial, la talla baja y las características cognitivas y de conducta, ya que la niña no tenía defectos cardíacos, ni hipercalcemia. En el caso 2, el niño tenía fenotipo facial y estenosis aórtica supravalvular, además de retraso del desarrollo sicomotor y carácter alegre y sociable. El niño también tenía reflujo gastroesofágico, cristales de oxalato de calcio en la orina y un FISH positivo para la deleción 7q11-23. En el caso 3 el diagnóstico se realizó por el fenotipo facial, la estenosis aórtica supravalvular y el retraso del desarrollo sicomotor. Como vemos, la característica mas importante para el diagnostico del síndrome de Williams es el fenotipo facial y las características cognitivas y de conducta, ya que la estenosis aórtica supravalvular y la hipercalcemia, así como los otros síntomas, no siempre están presentes y en nuestro país es difícil realizar los estudios específicos de diagnostico ya que no se hacen aquí y tienen un costo relativamente elevado. Existe una tabla de tamizaje clínico para el síndrome, de la Academia Americana de Pediatría y dependiendo del puntaje obtenido en la misma, se solicita el FISH para el cromosoma 7 o no16. Para realizar el diagnostico, en general, además de una historia clínica exhaustiva y examen físico se debe realizar; una ecocardiografía, calcemia, estudios tiroideos, ecografía abdominal y renal, evaluación neurológica, oftalmológica y auditiva4, 6. El manejo y tratamiento dependen de los síntomas presentes y los controles genéticos se realizan cada 6 meses en la infancia y anualmente desde el año en adelante. Se debe resaltar el hecho de que hay varios trabajos que reportan el fallecimiento de estos niños relacionado con la anestesia6. En general el SW ocurre en forma esporádica, pero debe realizarse un interrogatorio exhaustivo y examen clínico de los padres ya que se describen casos de transmisión de padres a hijos17. El síndrome de Williams es un síndrome relativamente común y que puede tener complicaciones serias, por lo tanto debe realizarse el diagnostico de certeza y en lo posible en forma precoz, para el mejor manejo del niño y para prevenir las posibles complicaciones.
BIBLIOGRAFIA
1. Jones KL. Smith's recognizable patterns Of human malformation. 5º ed. Philadelphia: W.B. Saunders Company; 1997. [ Links ]
2. Buyse ML. Birth defects encyclopedia. Center for Birth Defects Information Services. Massachusetts : Blackwell Scientific Publications; 1990. [ Links ]
3.London Dysmorphology Data Base. Neurogenetics. [base de datos en Internet]. London: Oxford University Press[acceso 7 de febrero de 2007]. Disponible en: http://www.amazon.ca/London-Dysmorphology-Database-Neurogenetics-combined/dp/0192686550. [ Links ]
4.Gorlin JR, Cohen MM, Hennekam RCM. Syndromes of the Head and the Neck. 4a ed. New York: Oxford University Press; 2001. [ Links ]
5.McKusick VA. On Line Mendelian Inheritance in Man. Omim. 194050. [base de datos en Internet]. USA: John Hopkins University Press. 2006. [acceso mayo de 2006]. Disponible en: http://www.ncbi.nlm.nih.gov/sites/entrez [ Links ]
6. Cassidy SB, Allanson JE. Management of genetic syndromes. 2º ed. New Jersey: Wiley-Liss;.2005. [ Links ]
7.Rimoin DL, Connor JM, Pyeritz RE. Emery and Rimoin's principles and practice of Medical Genetics. 3º ed. New York: Editorial Churchill Livingstone; 1997. [ Links ]
8.Antonell A, Del Campo M, Flores R, Campuzano V, Perez-Jurado LA. Williams syndrome: its clinical aspects and molecular bases. Rev Neurol 2006; 7(42 Suppl 1): 69-75. [ Links ]
9.Eronen M, Peippo M, Hiippala A, Raatikka M, Arvio M, Johansson R, et al. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet 2002 Aug; 39(8):554-8. [ Links ]
10.Farran EK. Orientation coding: a specific deficit in Williams syndrome? Dev Neuropsychol 2006; 29 (3): 397-414. [ Links ]
11.Farran EK, Jarrold C. Evidence for unusual spatial location coding in Williams syndrome: an explanation for the local bias in visuo-spatial constructuion tasks?. Brain Cogn 2005 Nov;59(2):159-72. [ Links ]
12.Greer MK, Brown III FR, Pai GS, Choudry SH, Klein AJ. Cognitive, Adaptive and Behavioural Characteristics of Williams syndrome. Am J Med Genet 1997. 74 (5): 521-5. [ Links ]
13.Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci 2006 May;7(5):380-93. [ Links ]
14.Gothelf D, Farber N, Raveh E, Apter A, Attias J. Hyperacusis in Williams syndrome. Characteristics and associated neuroaudiologic abnormalities. Neurology 2006 Feb 14; 66(3): 390-5. [ Links ]
15.Taybi H, Lachman RS. Radiology of syndromes, metabolic disorders and skeletal dysplasias. 4a ed. St Louis Missouri: Mosby; 1996. [ Links ]
16. Committee on Genetics. Health care Supervision for children with Williams Syndrome. Pediatrics 2001; 107:1192-204. [ Links ]
17.Thomas NS, Durkie M, Potts G, Sandford R, Van Zyl B, Youings S, et al. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11,23,15q11-q13 and 22q11. Eur J Hum Genet. 2006 Jul;14(7):831-7. [ Links ]
*Autor correspondiente:Dra. María Beatriz de Herreros
E-mail: mara@cmm.com.py

