











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La Fibrosis Quística (FQ) es una patología genética de patrón autosómico recesivo1. Se produce como consecuencia de mutaciones que afectan a un gen, el cual codifica para una proteína de membrana, cuya función es actuar como canal de cloro y se denomina Regulador de Conductancia Transmembrana de FQ o CFTR por sus siglas en inglés: Cystic Fibrosis Transmembrane Conductance Regulator2,3,4,5. El CFTR es esencial para la regulación de los movimientos de sal y agua a través de las membranas celulares3. La función ausente o reducida del CFTR da lugar a la producción de secreciones mucosas espesas en órganos con revestimiento de células epiteliales, que conducen a la obstrucción de los conductos que los trasportan4. El compromiso es multisistémico, pero afecta principalmente al aparato respiratorio, el sistema digestivo (6, las glándulas sudoríparas y el conducto deferente 7.
Aunque se describe que la FQ compromete principalmente a la población caucásica2) actualmente se reconoce que ningún grupo étnico puede ser considerado exento de FQ. Así, la incidencia puede variar desde 1/2.500 en Australia a 1/10.0000-35.0000 en Japón. En Latinoamérica las estimaciones sugieren que la FQ afecta entre 1/6.000-14.000 nacidos vivos8. En Paraguay, según datos preliminares, aún no publicados por el Programa Nacional de Detección Neonatal-Sub Programa Fibrosis Quística, la incidencia sería de 1/4.963.
En la FQ diferentes son los factores que favorecen un estado nutricional sub óptimo. Por un lado la insuficiencia pancreática, que puede conducir a mal absorción y por otro las infecciones respiratorias recurrentes con inflamación persistente de la vía aérea2 secundaria sobre todo a la colonización crónica por Pseudomona aeruginosa9. Consecuentemente a lo mencionado, un gasto energético incrementado sumado a una insuficiente ingesta alimentaria, se constituyen en aspectos determinantes para la instalación de un estado de desnutrición 2,4.
Sin embargo, diferentes registros internacionales de pacientes con FQ, como el de la European Cystic Fibrosis Society (ECFS)(10 o la Cistic Fibrosis Foundation (CFF) (11, los registros de Australia 12 o el Reino Unido 13, reportan una mejora en el estado nutricional durante las últimas 2 décadas, con incremento paralelo en la supervivencia. Esto, debido al manejo multidisplinario en centros de FQ, con el empleo de un soporte nutricional adecuado, en base al incremento del aporte energético (aumentando incluso hasta el 200% de los requerimientos para la edad en las fases de exacerbación respiratoria) 2,3,14). Todo lo mencionado, acompañado además de una buena adherencia a la suplementación enzimática, así como de vitaminas y minerales,15,16,17 junto a los avances terapéuticos para el mantenimiento de una mejor función pulmonar, y el diagnóstico precoz y oportuno a través de la pesquisa neonatal.
El objetivo del presente estudio, fue caracterizar el estado nutricional de los niños y los adolescentes con FQ, en seguimiento en el Programa Detección Neonatal-Sub Programa de Fibrosis Quística, en base a las guías y recomendaciones actuales.
MATERIAL Y MÉTODOS
Estudio observacional, descriptivo, con componente analítico, utilizando los datos de los registros médicos de los niños y adolescentes con FQ, que acuden al programa de Detección Neonatal-Sub Programa de Fibrosis Quística dependiente del MSPyBS. Para el mismo se consideró el control correspondiente a la revisión anual o semestral de aquellos pacientes menores de 19 años de edad, que consultaron entre febrero y noviembre del año 2016 y que al momento de la evaluación no presentaron exacerbación respiratoria 16,17.
Cada niño o adolescente fue identificado con un código, registrándose fecha de nacimiento, fecha de consulta, sexo, edad actual, procedencia, edad al diagnóstico, forma clínica de presentación, peso, talla/estatura, perímetro cefálico (este último en < de tres años de edad), valor del Volumen de Espiración Forzado en el primer segundo (VEF1), presencia o no de suficiencia pancreática y presencia o no de colonización crónica por Pseudomona aeruginosa, en cultivo de secreción traqueal.
Las mediciones antropométricas fueron realizadas por la autora principal, según protocolos estandarizados, siguiendo las directrices de evaluación nutricional del Instituto Nacional de Alimentación y Nutrición (INAN) del MSPyBS 18.
Los datos antropométricos fueron tipificados (puntaje z) según las curvas de referencia de la Organización Mundial de la Salud (OMS) 2006. En cuanto a la clasificación del estado nutricional, cada niño o adolescente se clasificó en riesgo de desnutrición, desnutrido, normal, sobrepeso u obesidad según los puntos de corte de la OMS 2006 19. Y se consideró como meta de estado nutricional óptimo en FQ, alcanzar, en niños menores de dos años de edad, una relación Peso/Talla (P/T) en 0 DE (Mediana) o percentil 50 (P50) y en niños iguales o mayores a dos años de edad, un Indice de Masa Corporal/Edad (IMC/Edad) en 0 DE (Mediana) percentil 50 (P50), pues esto se relaciona con una mejor función pulmonar. 2,20,21,22,23.
La suficiencia pancreática se evaluó midiendo el esteatocrito en muestra de heces, considerándose presencia de insuficiencia pancreática a aquellos con valores mayores al 2% 16.
La medición del VEF1, fue realizada a través de espirometría, en el control de rutina con neumología pediátrica, en niños de 6 años o más, definiéndose las siguientes categorías: Normal: VEF1≥ 80%, afectación leve 65-79%, afectación moderada 41-64% y afectación severa VEF1≤ 40 %. 21).
Los datos recolectados fueron digitalizados en una planilla de Microsoft Office® Excel® 2007 y posteriormente analizados con el software estadístico STATA® versión 12. Se evaluó la distribución normal de las variables mediante métodos de inspección visual y test estadísticos (Shapiro Wilk). A continuación se procedió a utilizar la estadística descriptiva para la presentación de los resultados. Las variables continuas fueron resumidas en medidas de tendencia central y dispersión, se utilizó promedios y desviación estándar cuando la distribución fue normal, y medianas y rangos intercuartílicos cuando la distribución fue no paramétrica. En el caso de las variables categóricas se realizaron cálculos de porcentaje y frecuencia absoluta por categoría.
Para explorar las diferencias entre promedios se utilizó la prueba estadística test t Student y ANOVA, en el caso de las variables categóricas se utilizó el Test exacto de Fisher y para observar la relación entre variables continuas se utilizó el test de correlación de Pearson, se consideró un nivel de significación estadística igual a p<0,05.
Para el cálculo de las puntuaciones estandarizadas de los indicadores zPeso/Edad, zPeso/Talla, zT/E, en niños < 5 años, se utilizó el programa Anthro de la OMS y para el cálculo de las puntuaciones estandarizadas de z IMC/E y z Talla/Edad, en los niños ≥ 5 años se utilizó el programa Anthro Plus de la OMS.
RESULTADOS
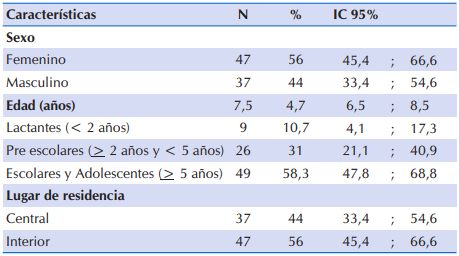
Fueron incluidas 84 fichas clínicas. Las características sociodemográficos de los niños y adolescentes con FQ se presentan en la Tabla 1. El 56% (n=47) correspondió al sexo femenino. En promedio, la edad fue de 7,5 ± 4,7 años (mínimo 1,2 años y máximo 18,6 años). Al agrupar en categorías por edad, el 10,7% fueron lactantes menores de dos años de edad, un tercio del total estuvo en el grupo comprendido de pre escolares y aproximadamente seis de cada 10 participantes tuvo más de 5 años de edad. En relación al lugar de residencia, el 56% (n=47) residía en el interior.
Tabla 1 Características sociodemográficas de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS (2016)
Valores expresados como media y desviación estándar [X̅ (DE)] (para todos los valores de este tipo)
La mediana de edad al diagnóstico fue de 4,9 meses, con un rango intercuartílico de 15,8 meses (p25=1,9 meses y p75=17,7 meses). En la Figura 1 se observa la distribución de los pacientes según la edad de diagnóstico. El 64,3% (n=54) de los niños fueron diagnosticados con FQ durante los primeros seis meses de vida.
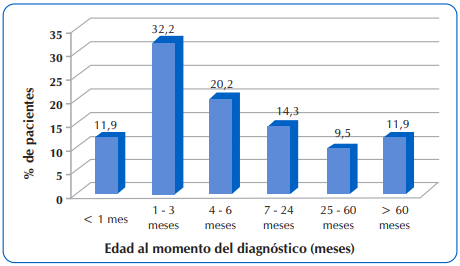
Figura 1 Distribución de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS, según la edad al momento del diagnóstico.
Las manifestaciones clínicas principales a la hora de la sospecha diagnóstica, fueron en el 41,7% de los casos, la denominada forma mixta, combinación de compromiso simultaneo, digestivo y respiratorio. Del 32,5% de los niños asintomáticos, (sin motivo de consulta clínica), en 22,6% (n=19) el diagnóstico se realizó por detección neonatal y en el 9,9% el diagnóstico se confirmó por datos de antecedentes familiares. Figura 2.
Con respecto a la presencia de insuficiencia pancreática, la misma se presentó en el 88% de los pacientes (n=74) IC: 81,2-95 (Datos no mostrados en tablas).
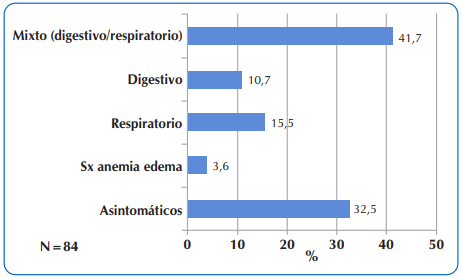
Figura 2 Distribución de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS, según las manifestaciones clínicas por Aparato y Sistema a la hora de sospecha diagnóstica
Se destaca en la Figura 3 el porcentaje de niños diagnosticados con FQ por el Test del piecito (detección o pesquisa neonatal) en relación a la población total de niños con dicha patología.

En la Tabla 2 se observan las características antropométricas de los niños y adolescentes con FQ, los mismos presentaron en promedio estaturas en el límite de lo considerado como normal, según la población de referencia de la OMS (zT/E=-1,0 DE), sin los promedios de la puntación z score del P/E, del P/T e IMC/E se encontraron dentro de lo esperado de acuerdo al estándar (z P/E: -0,6; z P/T: 0,5; z IMC/E:- 0,2). En los niños menores de tres años de edad, el perímetro cefálico para la edad (PC/E) se encontraba dentro de los valores adecuados según los estándares de referencia (z PC/E:-0,8).
Tabla 2 Características antropométricas de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS (2016).
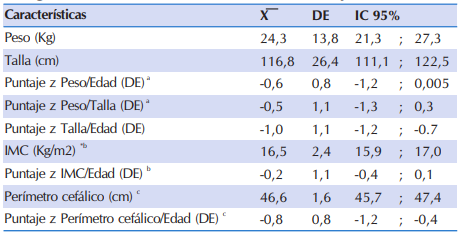
*IMC: Índice de Masa Corporal, DE: Desviación estándar
a Estimado en 9 niños < 2 años
b Estimado en 75 niños ≥ 2 años y < 19 años
c Estimado en 18 niños < 36 meses
En la Tabla 3 se observa la clasificación general del estado nutricional de los niños y adolescentes, según los indicadores de la OMS: P/E, PT, IMC/E, T/E y PC/E.
Tabla 3 Estado nutricional de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS (2016)

a Estimado en 9 niños < 2 años
b Estimado en 75 niños ≥ 2 años y < 19 años
Para una mayor ilustración gráfica, se observan en la Figura 4 los detalles del estado nutricional de niños con FQ menores de dos años de edad (n=9) y en la Figura 5 el resumen del estado nutricional de los niños con FQ mayores de dos años de edad.
Se destaca que en los menores de dos años de edad con FQ, ningún lactante presentó talla baja, el 56,6% presentó talla normal y un 11% presentó problemas relacionados a desnutrición moderada. Igual porcentaje presentó malnutrición por exceso (sobrepeso). Finalmente, al evaluar la prevalencia de microcefalia en los menores de 36 meses, ocho de cada 10 presentó perímetro cefálico adecuado, poco más del 10% presentó riesgo de microcefalia y el 11,1% restante presenta microcefalia. Figura 4.
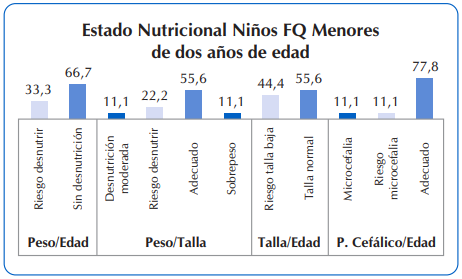
Figura 4 Estado nutricional de niños con FQ menores de dos años de edad, que asistieron al Sub Programa de Fibrosis Quística del MSPyBS (2016)
En el grupo de niños mayores de dos años y menores de 19 años (n=75), según el indicador IMC para la edad, seis de cada 10 niños tuvo un estado nutricional adecuado, uno de cada cuatro presentó riesgo de desnutrición, aproximadamente el 11% tiene sobrepeso y cerca del 3% obesidad, Al utilizar el indicador Talla para la edad para este grupo etáreo, el 17,3% presentó talla baja para la edad y poco más de un tercio riesgo de talla baja. Figura 5.

Figura 5 Estado nutricional de niños con FQ mayores de dos años de edad, que asistieron al Sub Programa de Fibrosis Quística del MSPyBS (2016)
Al comparar los datos antropométricos de los niños y adolescentes por sexo, se observó que si bien las mujeres presentaron menor puntaje z de P/E y T/E que los hombres, estas diferencias no alcanzaron a tener significancia estadística. Al clasificarlos por rango etario, entre los dos y 10 años se presentó el menor puntaje de T/E, sin embargo las diferencias por categorías de edad no fueron significativas, para ninguno de los indicadores (Tabla 4).
Tabla 4 Datos antropométricos de los niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS, clasificados según sexo y grupo de edad

a Diferencias por sexo evaluadas utilizando Test t-Student
b Estimado en 9 niños < 2 años
c Estimado en los 84 niños
d Estimado en 75 niños ≥ 2 años y < 19 años
e Diferencias por grupo etario evaluadas utilizando ANOVA
Al considerar la meta de estado nutricional en niños con FQ mayores de dos años de edad, de IMC/E igual o mayor a la mediana, se encontró que 32 niños y adolescentes (42,7%) estuvieron en este grupo. Sin embargo, también se destaca que 10 de los 32 niños presentó malnutrición por exceso. Figura 6.
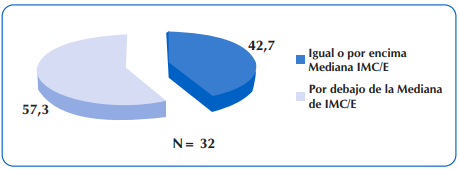
Figura 6 Porcentaje de niños y adolescentes con FQ mayores de dos años de edad, que presentaron IMC/E igual o mayor a la mediana.
En relación al aislamiento de Psudomona aeruginosa, un total de 35 niños (41,7%, IC 31,2-52,2) presentó colonización crónica, siendo el promedio de edad 9±4,1 años. (Datos no mostrados en tablas).
En la Tabla 5, se observa que al analizar la función pulmonar a través de la espirometría (VEF1) en los niños de seis años o más, se constató que cerca del 50% presentó afectación leve y aproximadamente 12% afectación moderada, los demás (41,9%) tuvieron función pulmonar normal.
Tabla 5 Función pulmonar de 43 niños y adolescentes ≥ 6 años con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS
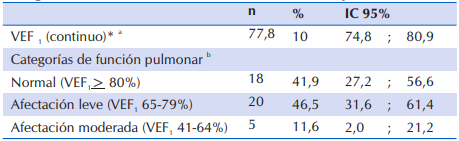
*VEF1: Volumen Espiratorio Forzado al Primer Segundo
a Valores expresados como media y desviación estándar [X̅ (DE)] (para todos los valores de este tipo)
b Estimado en 43 de 46 niños ≥ 6 años
Al comparar el puntaje z IMC según la ausencia o presencia de Pseudomona aeruginosa, se observó que los niños con presencia de Pseudomona aeruginosa tienen significativamente peor estado nutricional que aquellos que no presentan Pseudomona aeruginosa (promedio de z IMC 0,02 versus -0,5 DE; p =0,0324). Los datos constan en la Figura 7.

Figura 7 Comparación del estado nutricional de 84 niños y adolescentes con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS, según presencia o no de Pseudomona aeruginosa
En la Tabla 6 se observa que, los niños y adolescentes que se encontraban con desnutrición o en riesgo de padecerla, presentaron significativamente peor función pulmonar que aquellos que no estuvieron en esa condición.
Tabla 6 Diferencia promedio en el volumen espiratorio forzado al primer segundo en 43 niños y adolescentes ≥ 6 años con FQ que acudieron al Sub Programa de Fibrosis Quística del MSPyBS, según estado nutricional.
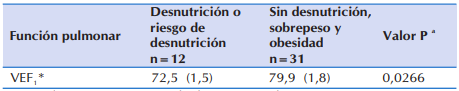
*VEF1: Volumen Espiratorio Forzado al Primer Segundo
a Diferencias por estado nutricional evaluadas utilizando Test t-Student
Al analizar la correlación entre la función pulmonar a través del VEF1 y el estado nutricional (z IMC) en 43 niños y adolescentes ≥ 6 años con FQ. se encontró un Coeficiente de correlación de Pearson: r = 0,2527 (p = 0,001). Figura 8.
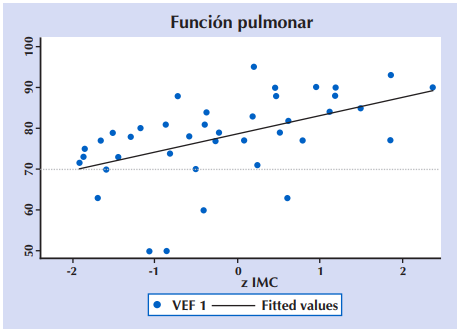
Figura 8 Correlación entre la función pulmonar (VEF1) y el estado nutricional (z IMC) en 43 niños y adolescentes ≥ seis años con FQ. VEF1: Volumen espiratorio forzado del primer segundo; z IMC: Puntuación z de índice de masa ponderal para la edad. Coeficiente de correlación de Pearson: r = 0,2527 (p = 0,001).
DISCUSIÓN
El déficit nutricional y el compromiso pulmonar progresivo son frecuentes en los pacientes con fibrosis quística. El deterioro clínico, asociado a altas tasas de mortalidad podría prevenirse con la detección neonatal y el diagnóstico precoz 24).
En el presente estudio, la mediana de edad al diagnóstico de FQ fue de 4,9 meses, siendo el 64% de los niños diagnosticados antes de los seis meses de vida, cifras que traducen un diagnóstico oportuno. En Paraguay, Garcete et al describieron en el año 2002 una mediana de edad al diagnóstico de 21 meses25 sobre una población de 31 pacientes, cifra que comparada con la actual investigación, evidencia un importante avance. Esta mejoría podría deberse a la creación, del Programa Nacional de Detección Neonatal en nuestro país, en el año 2005.
Al comparar la edad de diagnóstico de FQ con aquellos países que cuentan con registros nacionales de pacientes con FQ, se encontró que en Brasil la mediana de edad al diagnóstico es de 19 meses (26. En países desarrollados como Canadá, el reporte disponible hace referencia que el 66,5% 27 de los niños son diagnosticados antes del año de edad. El registro de EEUU comunica 3,8 meses como la mediana de diagnóstico11, Inglaterra 2 meses13, el registro europeo10) que abarca datos de 26 países informa 3,6 meses y finalmente en Australia la media es de 3 meses12) , gracias a los programas de detección neonatal que operan en todos sus estados. Así como Australia, el resto de los países del mundo, irá implementado de manera universal los programas de detección neonatal para FQ, y tal vez en unos años ya no hablemos de formas clínicas predominantes al momento del diagnóstico. De hecho hoy día, encontramos muy pocos trabajos actualizados entre cuyas variables se describen estos parámetros.
Se resalta por tanto la importancia de la detección neonatal para un diagnóstico y tratamiento oportuno. Las observaciones clínicas de las últimas décadas, muestran clara evidencia en cuanto a los beneficios referentes al diagnóstico en las primeras semanas de vida24,28,29,30, ya que ello posibilita el inicio precoz de la terapéutica específica, hecho que incide directamente en el pronóstico de la enfermedad. El cribaje en el recién nacido mejora el estado nutricional, previene la desnutrición, brinda oportunidades de realizar intervenciones precoces para reducir el daño pulmonar irreversible y es mucho menos costoso que un diagnóstico tradicional 31,32.
En el presente trabajo el 22% de los pacientes fue diagnosticado por prueba de detección neonatal, en comparación con Brasil y Argentina que reportan 22 y 29% respectivamente17,26. En países desarrollados el porcentaje es aún más elevado, llegando a 50% en Australia(29), 59,6% en EEUU11, 75% en la Unión Europea 10. En este sentido, debemos resaltar que la detección neonatal en Paraguay, se hizo universal para FQ recién a partir de enero del año 2016, motivo por el cual la cifra de diagnóstico sintomático sigue siendo aún la predominante.
En esta investigación la forma más frecuente de presentación clínica fue la mixta, una asociación de compromiso simultáneo, respiratorio y digestivo/nutricional, observado en poco más del 40% de los participantes. Este valor, si bien mantiene la tendencia, difiere a lo descripto en el año 201133 en el que el 68% de los pacientes evidenció una forma mixta de presentación al momento del diagnóstico. Este hecho podría atribuirse a que entonces el 100% de los diagnósticos fueron sintomáticos, no estando presentes aun la categoría de forma asintomática, que incluye a los pacientes de detección neonatal y a aquellos diagnosticados por antecedentes familiares. El reporte realizado por Chaustre34) en Venezuela, halló también una mayor frecuencia de la forma mixta de presentación e incluso con valores más altos que el descripto en este estudio, 76% de los casos. Sin embargo, González et al 35) reportan en España sólo un 18% de casos con forma mixta de presentación, mientras que en el Brasil un 64% de los pacientes debutó con clínica respiratoria exclusiva26. Estas discrepancias podrían atribuirse probablemente a diferencias en el origen genético de las diversas poblaciones de FQ, así como a la variabilidad en la distribución de las diversas mutaciones.
Referente a la frecuencia de pacientes suficientes pancreáticos, el hallazgo de esta investigación fue similar a la descripta en estudios internacionales de diferentes series 2,3,4,11).
La frecuencia de desnutrición en FQ es muy variable, con cifras que pueden oscilar entre el 10 y 50% o incluso más2,35 dependiendo de varios factores, entre los que podemos citar: la edad de diagnóstico, la detección por pesquisa neonatal, el criterio utilizado para su definición, tales como parámetros antropométricos, patrones de referencia locales o internacionales, así como los puntos de corte36. Dado lo trascendente de este punto y con la premisa de unificar criterios, en el año 2005 la CCF propone a un grupo de expertos reunirse con el objeto de evaluar criterios diagnósticos en FQ y a la vez definir los objetivos nutricionales20). El grupo de trabajo sugirió utilizar el IMC/E dada su mayor sensibilidad para detectar déficit nutricional y su estrecha correlación con la función pulmonar. El punto de corte recomendado como meta, fue mantener un IMC/E ≥ al P50 o ≥ a 0 DE en niños y adolescentes y una relación P/T ≥ al P50 o ≥ 0 DE en niños menores de 2 años de edad (2, 4, 20). Esta directriz se basa en que sobre la mediana o por encima del percentil P50 de IMC, el VEF1 evidencia un plateau superior a 80% y bajo la mediana o el P50 se deteriora, con mayor pendiente bajo P10 (20, 21, 22, 23).
En esta investigación, si bien el 74% de los pacientes presentaron un IMC en rango según criterios de la OMS, solo el 42,7% llegó a la meta u objetivo nutricional. Como antecedente del tema a nivel nacional, en el año 2014 en una investigación previa, se reportó un porcentaje del 50% de pacientes en rango para IMC, de los cuales solo un 27% 37 llegaba entonces a la meta u objetivo nutricional, constatándose que los resultados actuales evidencian mejoría en las características nutricionales de los pacientes en Paraguay. Esta situación es experimentada desde hace tiempo atrás en otros países, documentada a través del reporte periódico de sus registros. Así, en el correspondiente al de EEUU, se puede apreciar claramente como los pacientes entre dos y 20 años han incrementado en las últimas décadas su IMC entre 15 y 25 percentiles (2, 11).
La tendencia actual para comparar el estado nutricional entre diferentes países o regiones, es utilizar el valor de la media del puntaje z para IMC/E o su ubicación con respecto al P50. El valor de la media para IMC/E encontrada en esta investigación fue de -0,2 DE, que equivaldría en torno al P45. Al comparar estos datos con los registros internacionales de pacientes con FQ, son similares al de Brasil, que presenta una media de IMC en el P43, lo que equivaldría a que el 57 % de su población FQ no llega a la meta u objetivo nutricional 26). Para EEUU, la media se sitúa en el P5411) para Canadá en el P4427, Inglaterra P5413 y la Unión Europea P5010. Estos datos generales traducirían, que la adhesión al tratamiento enzimático y el empleo de un apoyo nutricional intensivo basados en dietas hipercalóricas, han hecho posible que en las últimas décadas la mejoría del estado nutricional sea realidad, con todas las ventajas que esto supone en cuanto a morbi mortalidad (2,4).
La talla baja es una observación frecuente y de causa multifactorial en los niños y adolescentes con FQ, estando asociada la mayoría de las veces al diagnóstico tardío o a desnutrición severa. El registro de EEUU reporta un 9,9% de talla baja11 entre sus pacientes, Chile 12%21, Brasil (26 en su registro no informa porcentajes, pero la media de z score para la talla es de -0,5, equivalente al P25, Australia 2%29, todos inferiores al 16% de talla baja reportada en este trabajo, situación que traduce un importante número de niños con desnutrición crónica, posible reflejo de marcado déficit ponderal en el pasado. Tienen importante influencia las características parentales de cada muestra, siendo una limitante en este caso no haber considerado o tenido en cuenta la talla genética para su corrección. Desde hace unos años se plantea, la necesidad de tal vez redefinir la talla baja en este grupo de pacientes y evaluar la necesidad de dar mayor énfasis a la velocidad de crecimiento 22.
Por otra parte, la obesidad y el sobrepeso en la edad infantil se han convertido en una auténtica epidemia tanto en los países desarrollados como en desarrollo. En Paraguay, la prevalencia de sobrepeso/obesidad en niños y adolescentes es cercana al 33% . Las causas de este fenómeno son multifactoriales, entre éstos, un mayor consumo de alimentos con alto contenido energético, la disminución de la actividad física y un mayor número de horas frente al televisor y computadoras. Esta tendencia en el estilo de vida no es exclusiva de la población sana, afectando a los pacientes con enfermedades crónicas, como la FQ, probablemente con mayor intensidad35. Es importante destacar que dos niños presentaron obesidad y ocho sobrepeso (en conjunto 13% del total). Este es un hallazgo bastante significativo ya que aparece en una patología históricamente relacionada con la desnutrición. En la literatura internacional, no existen datos relevantes sobre la verdadera frecuencia de este problema tal vez emergente en la FQ. Las publicaciones sobre el tema son muy escasas, de hecho los registros nacionales o regionales, no hacen ninguna mención al respecto, excepto el registro de Canadá27 que cita una frecuencia de 2% para obesidad y 7% para el sobrepeso. En Inglaterra se citan cifras del 10%28, en Grecia del 13,7%. 38 y en España del 5% 35.El sobrepeso puede influir negativamente en la función pulmonar, y a la vez si está presente favorecer la aparición de complicaciones asociadas tales como la diabetes relacionada a la FQ 39, las dislipidemias y la esteatosis hepática. Se considera por tanto, que el soporte nutricional debe ser individualizado. De hecho, desde hace unos años se viene planteando, de la necesidad real de mayor aporte energético en todos los casos. Postulándose a la fecha, que el niño debe recibir una alimentación balanceada teniendo como parámetros de evaluación de requerimientos energéticos, sus curvas de ganancia en peso y talla2). Si con un aporte energético común para un niño sano, el niño con FQ crece y se desarrolla normalmente, no ameritaría un aporte extra sino, una vigilancia cercana para detectar cualquier necesidad de intervención oportuna (2, 4,40.
La medición del perímetro o circunferencia craneana es fundamental hasta los 3 años de vida18 ya que experimentan un gran aumento debido al crecimiento del encéfalo y a la maduración del sistema nervioso central. Este es afectado por el estado nutricional, una menor circunferencia cefálica está asociada a mayores posibilidades de retardo mental. Cabe destacar que dos pacientes presentaron microcefalia y otros dos lactantes, riego de microcefalia. Esta situación podría corresponder a pacientes con antecedentes de marcada afectación nutricional en los primeros meses de vida. Muchos factores influyen para un adecuado control y la posibilidad de realizar intervenciones óptimas. También las frecuentes internaciones empeoran aún más el déficit nutricional, por las horas de ayuno y el tipo de alimentación recibida durante los ingresos hospitalarios. Estos pacientes se beneficiarían con un apoyo de estimulación oportuna. En este caso, los dos niños que presentaron microcefalia, tenían antecedente de ileo meconial, con numerosas re intervenciones y largas estadías hospitalarias.
La colonización crónica por Pseudomona aeruginosa9, se ha asociado con mayor deterioro de la función pulmonar y peor pronóstico más aún si ocurre durante los primeros años de vida. Los controles rutinarios, con cultivos de secreción traqueal cada 2 a 3 meses 16,17,41 según el caso en particular, permite el aislamiento precoz de esta bacteria, en fase asintomática42,43,44, permitiendo así iniciar de inmediato tratamiento agresivo, para intentar erradicación de la Pseudomona aeruginosa y de esa manera posponer el inicio de la colonización crónica16,17. En la edad adulta el porcentaje de colonización puede ser tan alto como del 70 a 80% 17,43.
En este estudio el 40% de los participantes mostró colonización crónica para dicho microorganismo, en comparación con lo reportado por Brasil que describe cifras del 37% 26, EEUU de 47% 11 y la Unión Europea de 30%. (10,44. El promedio de edad de colonización en el presente trabajo fue de 9,04 años, bastante precoz probablemente por antecedente de diagnóstico tardío, situación en que un alto porcentaje ya está colonizado desde el diagnóstico y generalmente presenta antecedentes de internaciones por cuadros respiratorios a repetición. A diferencia de lo reportado por Inglaterra, donde los niños de 9 a 11 años presentan colonización solo en el 27% de los casos44. En cambio EE UU llega a nuestra misma proporción con un 42% de colonizados11.
El menor IMC que se evidenció en los portadores de dicho microorganismo, coincide con lo descripto por Barja en Chile21. Dicha asociación se explica porque la colonización conlleva per se altos requerimientos energéticos, acentuados por la mayor frecuencia de exacerbaciones que producen además anorexia, disminuyendo la ingesta de alimentos y empeorando la desnutrición. 42,45
La correlación directa entre VEF1 e IMC también fue significativa en este estudio. Prácticamente todos los trabajos reportan este resultado. En la región, mencionamos a Barja et al. en Chile21 y la exhaustiva revisión de Marrichi et al. en Brasil que también lo confirman46. Aún no está claro el mecanismo de esta interacción, que probablemente depende de la preservación de masa magra, con mejor contractibilidad de los músculos respiratorios. Algunos estudios demuestran que la intervención nutricional puede asociarse a mejoría del VEF1 o al menos a desaceleración de su deterioro 45,46.
Varios estudios han demostrado que el IMC es un parámetro más sensible que el peso para la altura20. Sin embargo, el IMC no debe utilizarse de forma aislada para evaluar el estado nutricional de los pacientes con FQ, ya que no determina la composición corporal 2.
En la FQ, el desequilibrio entre la síntesis y la degradación proteica para satisfacer los requerimientos energéticos del cuerpo, puede conducir a una pérdida desproporcionada de masa corporal magra, con atrofia muscular, incluyendo atrofia muscular respiratoria. Dado que el peso corporal se compone principalmente de masa grasa y masa libre de grasa, la observación de estos dos compartimentos corporales por separado, puede proporcionar información más precisa sobre el estado nutricional, que está directamente relacionado con la enfermedad pulmonar, la principal causa de morbilidad y mortalidad en la FQ47.
En conclusión, dentro del manejo transdisciplinar de las personas con FQ, uno de los principales pilares lo constituye el apoyo nutricional, el cual deberá ser siempre personalizado. A la fecha, aun con los grandes avances terapéuticos para esta patología, todavía un grupo importante de pacientes presenta un estado nutricional sub óptimo. Pero por otro lado, el apoyo nutricional en algunos casos muy agresivo, sumado a un estilo de vida poco saludable, estaría generando, aunque todavía en pequeña proporción, el surgimiento de trastornos nutricionales por exceso.
A partir del presente trabajo, se plantea la necesidad de creación del Registro Nacional de FQ que haría posible el monitoreo continuo de la situación de todos los pacientes, de manera a disponer de datos que puedan orientar a toma de decisiones, con el objeto de mejorar la calidad de vida de los niños y adolescentes con FQ.

