








Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versión On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.8 n.1 Asunción jun. 2010
REPORTE DE CASOS
Anomalías cromosómicas en ambos padres de un niño con defectos congénitos múltiples. Reporte de caso
Chromosomal abnormalities in both parents of a child with multiple congenital defects. Case report
*Torres E, Herreros MB, Monjagata N, Rodríguez S, Sánchez S, Ayala A
Laboratorio de Genética. Departamento de Biología Molecular y Genética.
Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción (UNA). Paraguay
RESUMEN
Se presenta el caso de una pareja con antecedente de hijo portador de defectos congénitos, fallecido a los 2 días. No se realizó cariotipo al niño fallecido, quien es el producto del 3º embarazo de madre de 35 años y padre de 45 años, ambos aparentemente sanos y no consanguíneos. No refieren antecedentes familiares de defectos congénitos, no hay antecedentes patológicos ni de ingestión de medicamentos durante el embarazo.El estudio cromosómico de la pareja se realizó en linfocitos de sangre periférica. En todas las células analizadas de la madre se observó la heterocromatina del cromosoma 9 aumentada, en el 2% de las células, una translocación entre los cromosomas 2 y 22 y también en el 2% un isocromosoma para el brazo largo del cromosoma 1. En el padre, el 7,5% de las células analizadas, se observó la presencia de un cromosoma marcador.El cariotipo de la madre fue 46,XX,9qh+,t(2;22),i(1q); y del padre 46,XY/47,XY+mar.Si bien el 9qh+ es considerado variante cromosómica normal y sin repercusión clínica, esta condición ha sido hallada en parejas con problemas de esterilidad e infertilidad. La translocación y el isocromosoma hallados en la paciente, son alteraciones cromosómicas más severas, fueron observadas en muy bajo porcentaje, constituyen rearreglos cromosómicos que conducen a la formación de gametos desequilibrados. Sería más factible considerar al cromosoma marcador supernumerario como responsable de la malformación en el niño. Con estos antecedentes, se resalta la indicación médica del estudio citogenético en padres de pacientes portadores de malformaciones múltiples, para el asesoramiento genético familiar correspondiente.
Palabras clave: Defectos congénitos, cromosoma marcador, infertilidad.
ABSTRACT
This is the case of a couple with a child carrier of congenital defects, deceased at the second day after birth. The karyotype the deceased child was not made and the child was product of the third pregnancy of a mother of 35 years old and a father of 45 years old, both apparently healthy and non-consanguineous. Both parents did not refer any family history of birth defects, pathological background or medicines intake during pregnancy. The couples karyotype was performed on lymphocytes isolated from peripheral blood. All cells from the mother showed and increased heterochromatin on chromosome 9; 2% showed a translocation between chromosomes 2 and 22; and another 2% showed an isochromosome for the long arm of chromosome 1. In the father, 7.5% of the cells showed the presence of a marker chromosome. The mother´s kariotype was 46,XX,9hq+,t(2;22),i(1q) and the father´s was 46,XY/47,XY+mar. While 9qh+ chromosomal is considered normal with no clinical consequences, this condition has been found in couples with infertility and sterility problems. The translocation and the isochromosome found at a very low percentage are considered the most severe chromosomal abnormalities that lead to the production of unbalanced gametes. It would be more feasible to consider the supernumerary marker chromosome as responsible of the birth defects in the child. This background emphasizes the importance of the medical indication of cytogenetic studies in parents of children carriers of multiple malfomrations for the corresponding genetic counseling.
Keywords:Congenital defects, marker chromosome, infertility.
INTRODUCCION
El nacimiento de un niño con una malformación congénita lleva consigo la inevitable angustia de la familia y una sensación de culpabilidad. Es por ello que los padres buscan una explicación al problema. Según la OMS, los defectos congénitos son las anomalías del desarrollo morfológico estructural, funcional o molecular presentes al nacer, interna o externa, familiar o esporádica, hereditaria o no, única o múltiple.
Estas malformaciones ó defectos congénitos, se encuentran presentes al momento del nacimiento (1), y las causas son múltiples y variadas, siendo más de la mitad de los casos de causa desconocida, pueden ser multifactorales en un 25%, cromosómicas en un 10%, monogénicas en un 9% y en sólo 7% la causa es ambiental.
Teniendo en cuenta que los defectos congénitos afectan de un 5% a un 10% de los productos de los embarazos (2) y que muchos cuadros clínicos presentes en recién nacidos a término o prematuros son ocasionados por anomalías cromosómicas, varios autores han encaminado sus investigaciones en conocer la participación de estas anomalías en la etiología del aborto espontáneo y del recién nacido, los estudios cromosómicos se han realizado directamente en los productos abortados, en parejas infértiles y estériles y en recién nacidos (3-5). Se ha demostrado que la presencia de las alteraciones cromosómicas en las parejas infértiles ó estériles, son más frecuentes que en la población general como parte del proceso natural de evolución, cuando las alteraciones cromosómicas coexisten en las células germinales pueden transmitirse a los gametos y a la descendencia, en otros casos conducen a un arresto en la gametogénesis ó al desarrollo de gametos con alteraciones cromosómicas no balanceadas. Estas anomalías cromosómicas pueden ser numéricas o estructurales y pueden afectar a uno o más autosomas, cromosomas sexuales o ambos de manera simultánea. En un trabajo realizado en nuestro laboratorio, la prevalencia hallada en parejas con esterilidad e infertilidad fue de 5,7%, siendo las más frecuentes las variantes cromosómicas en un 52% de la población estudiada, seguida de anomalías numéricas en un 38% y las estructurales en un 9% (6,7).
CASO CLÍNICO
Se presenta el caso de una pareja que consulta por antecedente de un hijo fallecido a los 2 días, portador de defectos congénitos, entre los que se pueden mencionar, labio y paladar hendido unilateral, hipertelorismo, puente nasal chato y orejas de implantación baja. Por ecocardiografia mostró cardiopatía congénita (CIV) y (CIA), por ecoencefalografia mostró cisterna magna aumentada de tamaño con hipoplasia de vermis cerebeloso. El cordón umbilical presentó arteria umbilical única, con dos luces. Entre las anomalías de miembros presentó hipoplasia de radio y cúbito izquierdo, agenesia de cúbito derecho, ambas manos con 4 dedos y en posición bot. Sindactilia en los 2º y 3º ortejos y talones prominentes. Los genitales con hipospadias y criptorquidia bilateral. Uréter proximal derecho dilatado. No se realizó cariotipo al niño. La pareja consulta luego del fallecimiento del mismo con su historia clínica y fotos, solicitando asesoramiento genético. Por las características clínicas del hijo y el comprometimiento multisistémico, se sospecha que haya sido portador de anomalía cromosómica y se indica estudio cromosómico de ambos padres.
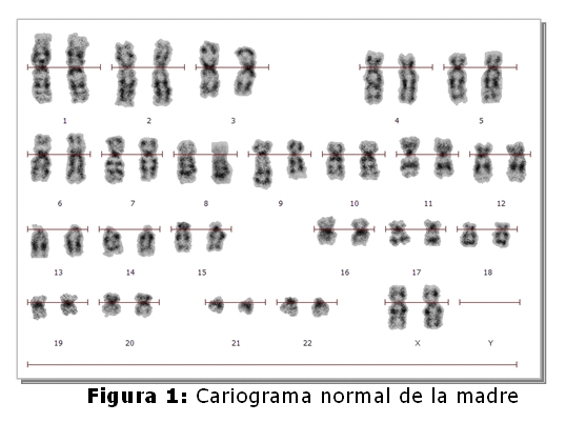
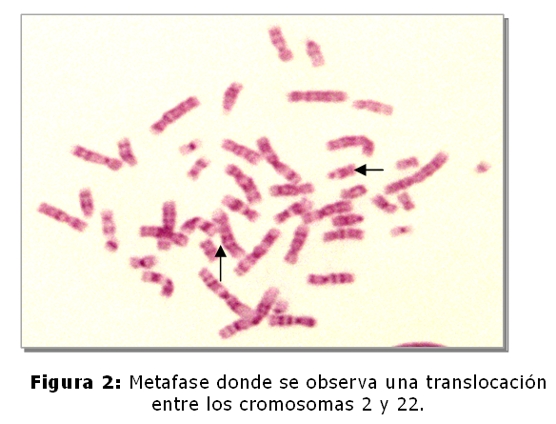
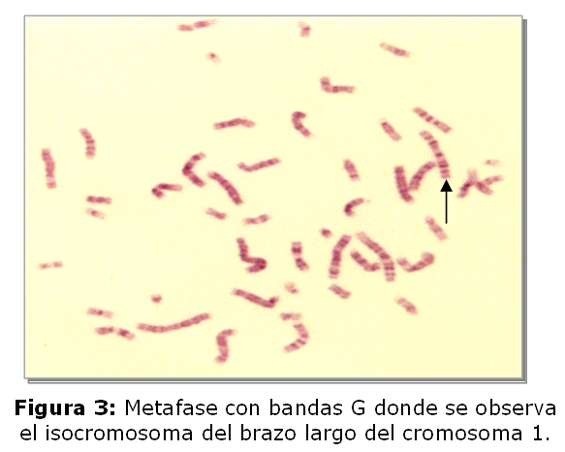
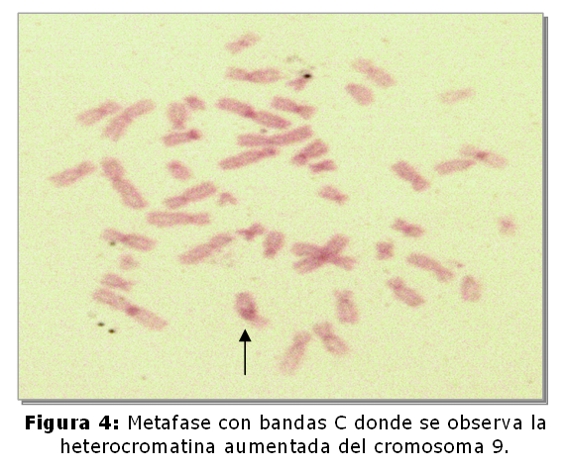
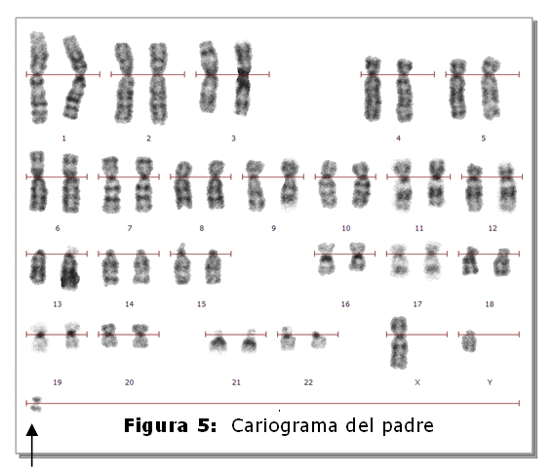
El niño fallecido es el producto del 3º embarazo de madre de 35 años y padre de 45 años, ambos sanos y no consanguíneos. Tienen dos hijos sanos, una niña de 9 años y un varón de 5 años. No refieren antecedentes familiares de defectos congénitos, no hay antecedentes patológicos ni de ingestión de medicamentos durante el embarazo. El estudio cromosómico de la pareja se realizó en linfocitos de sangre periférica. En todas las metafases analizadas de la madre se observó la heterocromatina del cromosoma 9 aumentada (Fig.1,4), en el 2% de las células una translocación entre los cromosomas 2 y 22 (fig.2) y en el 2% un isocromosoma para el brazo largo del cromosoma 1 (Fig.3). En el padre, el 7,5% de las células analizadas, se observó la presencia de un cromosoma marcador (Fig.5).
Cariotipo de la pareja: Madre: 46,XX,9qh+,t(2p;22q),i(1q).
Padre: 46,XY/47,XY+mar
DISCUSION
La heterocromatina aumentada del cromosoma 9 (qh+), es considerada una variante polimórfica normal (5), actualmente con refinadas técnicas moleculares se piensa que los genes para la fertilidad residen en la heterocromatina, el análisis de la secuencia del ADN del cromosoma 9 ha demostrado que es altamente polimórfico estructuralmente, con muchas duplicaciones intra e intercromosómicas, y contiene el mayor bloque autosómico de heterocromatina (5,8,9). En cuanto a la translocación y al isocromosoma hallados en la paciente, son alteraciones cromosómicas estructurales más severas, si bien éstas fueron observadas en muy bajo porcentaje constituyen rearreglos cromosómicos que conducen a la formación de gametos desequilibrados. Las translocaciones entre dos cromosomas autosómicos se han correlacionado con trastornos en la espermatogénesis, dando lugar a translocaciones balanceadas y no balanceadas, ambas asociadas con infertilidad, esto sugiere que estas modificaciones cromosómicas pueden causar errores en las secuencias promotoras o represoras de múltiples genes, lo que explica el amplio rango en el fenotipo(10). En relación al cromosoma marcador supernumerario hallado en el padre, con las técnicas utilizadas en el laboratorio no fue posible determinar el origen del mismo, para llevar a cabo su identificación debe realizarse en el paciente el estudio cromosómico con técnicas citogenéticas moleculares; sin embargo se puede considerar a este cromosoma marcador derivado de los brazos cortos de cromosomas acrocéntricos y sin relevancia clínica, sería el responsable por una segregación meiótica desbalanceada, la presencia de las malformaciones congénitas en el recién nacido (5,6).
Con estos antecedentes, se resalta la necesidad de la indicación médica del estudio citogenético en padres de pacientes con defectos congénitos múltiples, con el propósito de identificar alteraciones cromosómicas relacionadas con parejas con infertilidad y padecimientos que pudieran afectar a la descendencia y al embarazo, para su asesoramiento genético familiar.
REFERENCIAS BIBLIOGRÁFICAS
1. Aviña Fierro JA, Tastekin A. Malformaciones congénitas: clasificación y bases morfogénicas. Revista Mexicana de Pediatría 2008; 75(2):71-4. [ Links ]
2. Castilla E, López-Camelo J, Paz J, Orioli L. Los defectos congénitos y su prevención. En: Castilla E (ed). Prevención primaria de los Defectos Congénitos. Rio de Janeiro: Editora Fiocruz. 1996. [ Links ]
3.Van Assche E, Bonduelle MH, Tournaye H, Joris H, Verheyen GP, Devroey P et al. Citogenética de los hombres infértiles. Hum Reprod 1996;11(4):1-26. [ Links ]
4. Vega Conejo V, Viñas Portilla C, Lantigua Cruz A, del Monte Sotolongo E. Defectos cromosómicos y fallas reproductivas. Un estudio en 452 pacientes. Rev Cubana Obst Ginecol 1999; 25(1):19-23. [ Links ]
5. Torres E, Ascurra M, Rodríguez S, de Cabral M. Prevalencia de anomalías cromosómicas en parejas con trastornos reproductivos en Paraguay. Revista Iberoamericana de Medicina Fetal y Perinatal 2007; 1(3):142-7. [ Links ]
6. Torres E, Herreros MB, Rodríguez S, Ascurra M, Monjagata N. Monosomía 11q compatible con Sindrome de Jacobsen. Reporte de caso. Mem Inst Investig Cienc Salud 2006; 4(1):39-42. [ Links ]
7. Romero Tovar S, Juárez Espinosa B, Galindo García CG, Mendoza Romo M, Sánchez Usabiaga RA.Prevalencia de alteraciones cromosómicas en pacientes infértiles estudiadas en una clínica de reproducción asistida. Ginecol Obstet Mex 2009; 77(3):128-35. [ Links ]
8. Madon P, Athalye, Parikh F. Polimorphic variants on chromosomes probably play a significant role in infertility. Reprod Biomed Onl 2005; 11(16):726-32. [ Links ]
9. Krausz C, Giachini C. Genetic risk factors in male intertility. Arch Androl J Reprod Syst 2007; 53:125-33. [ Links ]
10. Behre HM, Nieschlag E, Horst J. Chromosomes abnormalities in 447 couples undergoing intracytoplasmatic sperm injection prevalence, types, sex distribution and reproductive relevance. Hum Reprod 1998; 13:576-82. [ Links ]
*Autor Correspondiente: Lic. Elodia Torres, Lab. de Genética. Departamento de Biología Molecular y Genética. Instituto de Investigaciones en Ciencias de la Salud. Río de la Plata y Lagerenza. Asunción-Paraguay
Email: genetica@iics.una.py
Fecha de recepción: mayo de 2010, Fecha de aceptación: junio de 2010