









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La hipertensión arterial pulmonar (HAP) es una patología derivada de la alteración de la vasculatura pulmonar que puede caracterizarse por proliferación o remodelación de los circuitos ya existente, y esto a su vez puede aumentar las resistencias vasculares pulmonares y llevar a un mayor esfuerzo cardíaco para lograr expulsar la sangre hacia estos vasos. Se define por cateterismo cardiaco derecho (CCD) cuando existe una presión media de la arteria pulmonar mayor a 25mmHg en reposo, con una presión capilar pulmonar <15mmmHg y con resistencias vasculares pulmonares > 2 o 3 unidades de Wood. Por ecocardiograma se le considera hipertensión a partir de una presión sistólica de la arteria pulmonar >40mmHg y se puede clasificar como leve cuando va de 40 a 54mmHg, moderada cuando va de 55 a 64mmHg y grave cuando es mayor a 64mmHg. (1-3).
La HAP es una condición hemodinámica que se produce por varias etiologías, que difieren en los niños respecto a los adultos, no solo por la edad de presentación, sino también en su incidencia y pronóstico. Siendo una característica de la edad pediátrica la etiología multifactorial(4).
La HAP incluye dos grandes grupos, los cuales la conforman como una misma definición, pero de origen fisiológico y fisiopatológico diferentes, estas son la hipertensión arterial pulmonar primaria o también conocida como idiopática, y la hipertensión arterial secundaria que se ve relacionada a otras patologías como lo son las enfermedades del tejido conectivo, entre otras (2,4).
Las enfermedades autoinmunes del tejido conectivo son un grupo heterogéneo de transtornos inflamatorios sistémicos que se caracterizan por la presencia de auto anticuerpos circulantes que afectan múltiples órganos y sistemas, dicho conjunto se correlaciona con frecuencia a la producción de HAP, dentro de este conjunto forma parte la esclerosis sistémica (ES) y las miopatías inflamatorias como lo son dermatomiositis, la polimiositis y la miositis por cuerpos de inclusión(5-6). Su diagnóstico es principalmente clínico y se apoya en hallazgos de laboratorio, exploración complementaria y abordaje multidisciplinario. Actualmente no existen criterios específicos para el diagnóstico de esclerosis sistémica en población pediátrica, sin embargo se han desarrollado varios criterios a través de la historia. En 1980, la clasificación inicial fue propuesta por el colegio americano de reumatología (ACR por sus siglas en inglés)(7). Posteriormente en 2007, se dió por primera vez el esfuerzo por la sociedad de reumatología pediátrica europea (PReS, por sus siglas en inglés) en conjunto con ACR, de proponer criterios para clasificar dichas patologías, los cuales tuvieron buena aceptación para el diagnóstico(8). En el 2013, la clasificación fue revisada nuevamente por la liga europea contra el reumatismo (EULAR), desarrollando un esquema de diagnóstico basado en puntajes(9), este último se encuentra en validación para población pediátrica, hasta el momento se ha reportado mejor desempeño frente a los criterios de PReS/ACR. En relación al diagnóstico de la dermatomiositis juvenil (DMJ), se basa en los criterios de Bohan y Peter(10), y de manera reciente, los criterios ACR/EULAR publicados en 2017, que permiten el diagnóstico de DMJ con o sin biopsia (11).
Una de las principales causas de disminución en la expectativa de vida en pacientes con enfermedades del tejido conectivo, como lo son la DMJ y ES, es la presencia de HAP, de modo que tienen una esperanza de vida menor al promedio para su edad y género.
El objetivo de nuestro estudio fue identificar la presencia de la HAP en pacientes con DMJ y ES en la población pediátrica de nuestro hospital, el cual representa un centro de referencia para el centro y sur del país.
MATERIALES Y METODOS
Estudio retrospectivo, descriptivo y transversal. La población de estudio incluyó a todos los pacientes pediátricos del Hospital para el Niño Poblano con diagnóstico de dermatomiositis juvenil o esclerosis sistémica entre el periodo comprendido del mes de octubre del año 2001-2023. Se excluyó a todo aquel paciente en el cual no fue posible concluir el diagnóstico de interés y aquellos que contaban con diagnósticos de sobreposición. El diagnóstico de dermatomiositis juvenil se realizó con base en los criterios de Bohan y Peter, y el diagnóstico de esclerosis sistémica con base en los criterios de PReS/ACR del 2007. Se realizó la revisión de los expedientes electrónicos por medio del sistema digital de nuestro hospital. Se recabó del expediente clínico la presencia de al menos un ecocardiograma con medición de la insuficiencia tricuspídea (IT) con la cual se estima la presión sistólica del ventrículo derecho (PSVD), que nos representa la presión sistólica de la arteria pulmonar (PSAP), siempre que no exista obstrucción hacia la salida del flujo pulmonar por el tracto de salida del ventrículo derecho y/o la arteria pulmonar, considerando la presencia o no de una presión elevada de la misma con umbral de 40mmHg. Se realizó el análisis descriptivo de las variables de interés, describiendo las medidas de tendencia central.
Algunas otras enfermedades del tejido conectivo han sido asociadas a HAP (Lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, enfermedad de Sjögren, etc.), sin embargo, no fueron contempladas para el estudio actual.
Se mantuvo la confidencialidad de los datos y se respetó los principios establecidos por la Ley General de Salud de nuestro país. De igual manera, el presente estudio se ajustó a los lineamientos de las convenciones de Helsinki y Tokio respecto a la confidencialidad de los participantes en el estudio. El presente estudio es retrospectivo, por lo tanto, no se firmó hoja de consentimiento informado, representa riesgo mínimo ya que no se realizó otras pruebas fuera de las solicitadas por los servicios tratantes.
Se contó con registro y aprobación por el comité de ética de nuestro hospital (HNP 2022-13).
RESULTADOS
Se encontró un total de 58 pacientes durante el periodo del 01 de octubre de 2000 hasta enero 2023, de los cuales la relación entre hombre mujer fue de 1:2.5, el diagnóstico de dermatomiositis estuvo presente en 21 pacientes mientras que el resto (37 pacientes) contaban con diagnóstico de esclerosis sistémica. La edad predominante al diagnóstico presentó dos picos, a los 6 y los 13 años, para ambas enfermedades.
Del total de nuestra población, se encontró valoración por cardiología en 17 pacientes (29,3%), dentro de los cuales, 10 contaban con diagnóstico de dermatomiositis juvenil y 7 con diagnóstico de esclerosis sistémica, 1 de cada grupo presentó hipertensión arterial pulmonar, definida por presión sistólica de la arteria pulmonar de 40mmHg o más, estimada por insuficiencia tricúspidea (velocidad >2.8m/s con presión de atrio derecho estimado de 10mmHg) por Ecocardiograma. En ningún caso se solicitó cateterismo cardiaco diagnóstico ya que la HAP encontrada fue leve sin necesidad de manejo farmacológico por el servicio de cardiología. Con base en lo previo, se obtuvo que el 10% de los pacientes con dermatomiositis juvenil presentó hipertensión arterial pulmonar al diagnóstico, y un 7% de los pacientes con esclerosis sistémica presentó HAP al debut (Figura 1).
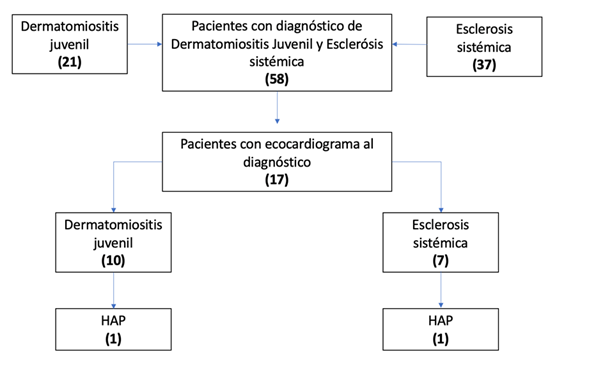
Entre los dos pacientes con HAP, uno contaba con diagnóstico de dermatomiositis juvenil y uno con esclerosis sistémica. La paciente con DMJ presentó una presión pulmonar en el límite inferior para clasificarse como hipertensión arterial pulmonar, y la paciente con ES presentó una presión pulmonar de 50mmHg. Ambas del género femenino, de 10 y 17 años respectivamente (Tabla 1). Ninguno de los pacientes fue sometido a cateterismo cardiaco por considerarse HAP leve.
Tabla 1 Pacientes con hipertensión arterial pulmonar.n:2.
| Sexo | Edad (años) | Diagnóstico | Presión Pulmonar Sistólica estimada | Datos clínicos* | Capilaroscopia | Perfil inmunológico |
|---|---|---|---|---|---|---|
| Femenino | 10 | Dermatomiositis | 40mmHg | Asintomática | Disminución en densidad capilar, capilares tortuosos generalizados. | ANA Positivos Anti Mi2 positivo Anti Ku positivo |
| Femenino | 17 | Esclerodermia | 50mmHg | Asintomática | S/D | S/D |
*Datos clínicos asociados a hipertensión arterial pulmonar. SD: Sin datos.
DISCUSIÓN
Dentro de la literatura histórica, Dowling consideraba que la DMJ y la ES pertenecían a un rubro único de enfermedad, por algunas de las características clínicas equivalentes que se presentaban, cuyo entendimiento actual permite separarlas. Si bien, es reconocido que ambos trastornos dentro de su efecto fisiopatológico pueden generar cambios degenerativos y oclusivos en las ramas pequeñas del árbol arterial pulmonar desembocando en hipertensión pulmonar, y de forma progresiva en la hipertrofia del corazón derecho, la asociación de HAP con DMJ es menos apreciada y sus características continúan siendo un rubro poco explorado, lo que resulta más notorio cuando se intenta encontrar estudios sobre la relación de dichas patologías(12-13).
En la literatura Internacional, en el 2001 se publicó en la British Society for Rheumatology por parte por Trapani y colaboradores(14), un estudio longitudinal donde se expresaba el involucro pulmonar en 12 pacientes con diagnóstico de DMJ, donde las medidas de capacidad pulmonar se obtuvieron por medio de espirometría, en el cual se tomaron en cuenta parámetros que involucraban las vías respiratorias bajas y la interfase alveolo-capilar por medio de los flujos máximos al 50% y 25% (MEF 50 Y 25) y la capacidad de difusión del monóxido de carbono (DLCO), la reducción de estos parámetros se manifestaron en dos pacientes, donde se observaba una hipertensión pulmonar secundaria a las anormalidades en la anatomía pulmonar y vascular.
En relación a los antecedentes nacionales, el estudio retrospectivo realizado por Villareal, en el Hospital Infantil de México “Federico Gómez” en el servicio de Reumatología pediátrica en el periodo de Enero de 2014 a Mayo de 2016, en una cohorte de 41 pacientes con diagnóstico de DMJ donde se encontró un predominio en el sexo femenino, se concluyó que la afección pulmonar inicial se documentó en 12,1% (n=5), donde el primer síntoma que se refirió fue la disnea. El 20% de los pacientes con afectación pulmonar presentaban HAP leve de 40 mmHg.(15)
Algunos anticuerpos han sido asociados con el desarrollo de afectación pulmonar o cardiaca en DMJ, específicamente los anticuerpos Anti-PM-Scl, Anti-Ku, Anti-MDA5, Anti-Mi2 y MRS se han asociado con el desarrollo de enfermedad pulmonar intersticial y la afectación cardiaca. Nuestra paciente presentó Anti-Mi1 y Anti-Ku positivo(16).
Por otro lado, en relación a los pacientes con ES, las estimaciones de la prevalencia de HAP en esta población oscilan entre el 6,4 % y el 9 % y representa una de las principales causas de morbilidad y mortalidad(17-19) La ecocardiografía trans-torácica es una herramienta de evaluación recomendada para la detección de HAP en pacientes con ES. Una velocidad Doppler de insuficiencia de la válvula tricúspide > 2,8 m/s se considera sospechosa de la presencia de HAP(20). En un estudio descriptivo transversal de 2010-2013 en población de Argelia con esclerodermia difusa y limitada, realizado por Nadera M. et, donde se incluyeron 202 pacientes (177 mujeres y 25 hombres), evaluaron la prevalencia de HAP, basandose en la velocidad máxima de insuficiencia tricuspídea (VIT) y disnea. Definiendo pacientes de alto riesgo (VIT > 3m/s o VIT entre 2,8 y 3m/s con disnea no explicada por otra causa), quienes fueron sometidos a cateterismo cardíaco derecho (CCD) para confirmar HAP. La sospecha de hipertensión pulmonar por ecocardiografía fue confirmada por CCD en el 68,2% de los casos, donde la VIT≥3,48m/s sugiere fuertemente HAP, resultando una prevalencia del 6%. Con un retraso medio de aparición de 7,08 años después del primer diagnóstico de ES. La HAP ocurrió en los primeros cinco años en el 58,3% de los casos y después de cinco años en el 41,6% de los casos, donde hubo mayor frecuencia en las formas cutáneas limitadas con 58,3%(19). Dichos resultados que coinciden con el estudio multicéntrico y metaanálisis realizado por Avouc J et al. (20), en 11 centros franceses e italianos donde se reclutaron 206 pacientes con ES en los que se sospechó HAP, de los cuales se confirmó por CCD en 83 pacientes (7%). El metanálisis identificó factores de riesgo para esta afección: la edad avanzada, la mayor duración de la enfermedad y el subconjunto de enfermedad cutánea limitada; en relación a la supervivencia, Lefèvre et al. observaron tasas de 1 y 3 años de 81 % y 52%, respectivamente, en un metanálisis(21). Si bien los niños con ES suelen tener mejores resultados en comparación con los adultos debido a la menor frecuencia de afectación orgánica grave, la HAP sigue siendo una de las principales causas de morbilidad y mortalidad en pacientes con ES(21).
A pesar de los avances en el tratamiento de la HAP, el pronóstico de HAP en ES es peor que otras formas de HAP con una mortalidad al año de hasta el 30%. En un informe francés reciente, durante el período 2006-2017, la supervivencia mejoró con el tiempo en pacientes de 70 años, pero no en pacientes mayores con ES e HAP(22). En un metaanálisis de nueve estudios, que incluyeron 2700 pacientes con ES, el 55% murió de causas relacionadas con ES. Entre estos, el 35% estaban relacionados con fibrosis pulmonar, 26% a HAP, 26% a insuficiencia cardiaca y arritmias y 4% a la crisis renal(23).
La HAP puede ocurrir en la ES secundaria a enfermedad intersticial pulmonar o enfermedad cardiaca, siendo más frecuente en la ES de tipo limitada y asociada fuertemente a los anticuerpos anticentromero. También se ha observado más frecuentemente en pacientes con fenómeno de Raynaud(24).
En México, no existen datos epidemiológicos con verdadero impacto acerca de la correlación entre la HAP con DMJ y ES; únicamente hay trabajos de tesis que hablan sobre su presencia en casos aislados (hipertensión arterial pulmonar leve). En este estudio se encontró que solamente el 29,3% tenían una ecocardiografía en algún punto del seguimiento hospitalario, por otra parte, la presencia de hipertensión arterial pulmonar se observó en un 11% de los pacientes que si tenían una ecocardiografía.
Dentro de nuestra población un 10% presentó hipertensión arterial pulmonar al diagnóstico de dermatomiositis juvenil, lo cual es más bajo en comparación con el estudio reportado por Villarreal, sin embargo, otros estudios no han reportado HAP en pacientes con DMJ(23). Por otra parte, el 7% de nuestros pacientes con ES presentó hipertensión arterial pulmonar al debut, lo cual es congruente con el resto de los estudios reportados. Si bien la baja presentación de la hipertensión arterial pulmonar en el grupo de los pacientes con DMJ puede estar influida por muchos factores, como lo pueden ser, la ausencia de ecocardiografía al diagnóstico, el tiempo de evolución de la enfermedad, el tratamiento previo o el fenotipo inmunológico, es difícil establecer en este momento un factor de riesgo o protector para el bajo porcentaje de HAP en nuestra población. En algunos casos se ha descrito el desarrollo de HAP de manera secundaria a la neumopatía intersticial, sin embargo, es controversial. En el caso de nuestra paciente, no presentaba neumopatía intersticial.
Una debilidad considerable del estudio que realizamos fue la ausencia de ecocardiograma en una porción importante de nuestros pacientes, así como la ausencia en el seguimiento de nuestros pacientes con estudios ecocardiográficos. En estudios posteriores se planea compensar estas debilidades.
CONCLUSIÓN
El presente estudio demuestra que la hipertensión arterial pulmonar se asoció en un 10% de los pacientes con dermatomiositis juvenil, y en el 7% con esclerosis sistémica, lo cual representa un bajo porcentaje para DMJ y mantiene el porcentaje reportado por otros estudios para la ES.
Este estudio pone en manifiesto, que continua habiendo una gran variación en el abordaje integral de las manifestaciones secundarias en las enfermedades del tejido conectivo en la unidad, habiendo pocos pacientes con un ecocardiograma en algún momento del curso de la enfermedad, desconociendo si la HAP se encuentra presente en el resto de las pacientes. Por lo cual, el protocolo de estudio en el momento del diagnóstico de dermatomiositis juvenil o esclerosis sistémica requiere una valoración cardiológica de base y un seguimiento constante.