









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El Síndrome de Wolf-Hirschhorn (SWH) es una enfermedad genética polimalformativa, también conocida como Síndrome 4p-, debido a la microdeleción en la región distal del brazo corto del cromosoma 4, específicamente por la pérdida de genes contiguos localizados en 4p16.31. Fue descrito por primera vez en 1.961 por Cooper y Hirschhorn, posteriormente, en 1965 por Wolf et al. La frecuencia de presentación de este Síndrome es de 1 en 20.000 a 50.000 nacidos vivos, presentando el doble de frecuencia en mujeres que en varones2.
Los pacientes con SWH se caracterizan por tener un fenotipo facial peculiar y rasgos craneofaciales típicos en la infancia que consiste en la apariencia de "casco de guerrero griego" de la nariz (puente ancho de la nariz que continúa hasta la frente, cejas arqueadas, hipertelorismo, filtrum corto y micrognatia), discapacidad intelectual, retraso del crecimiento intrauterino y posnatal, retraso del desarrollo psicomotor, epilepsia, dificultades de alimentación, defectos cardíacos congénitos, pérdida de audición, anomalías esqueléticas, malformaciones renales y del tracto urinario y anomalías oftalmológicas y dentales3.
En relación a la porción del cromosoma 4 que se selecciona en estos pacientes, reportes sobre esta enfermedad, sugieren que el tamaño de la región seleccionada varía entre las personas afectadas, las deleciones más grandes tienden a provocar discapacidades intelectuales y características físicas más graves, presentan una variabilidad fenotípica en función a la gravedad. Los reordenamientos que involucran al brazo corto del cromosoma 4 y que son de novo, ocurren con mayor frecuencia en la meiosis paterna, supone en gran medida a deleciones aisladas, sin embargo, se han descrito pacientes de novo portadores de translocaciones desequilibradas entre los brazos cortos de los cromosomas 4 y 8 con alta frecuencia y de origen materno4. La deleción 4p es posible detectar por citogenética convencional en asociación con el fenotipo característico, pero la microdeleción cromosómica solo es factible detectar a través de la técnica de Hibridación in situ por fluorescencia (FISH), que se utiliza para la confirmación del diagnóstico, facilitando de esa manera el tratamiento y lograr una mejor esperanza de vida de los pacientes, es por eso que el FISH se ha convertido en una herramienta importante para detectar y controlar una terapia específica para este tipo de enfermedad genética5.
En la Argentina, alrededor del 80% de los casos reportados son esporádicos y el 20% se originan a partir de un rearreglo familiar balanceado (Medina, Rojas, Guevara, Cañizales, & Jaimes, 2008). Este trabajo tiene como propósito describir el caso de una niña de 7 meses de vida con sospecha clínica de ser portadora del Síndrome de Wolf-Hirschhorn.
DESCRIPCIÓN DEL CASO CLÍNICO
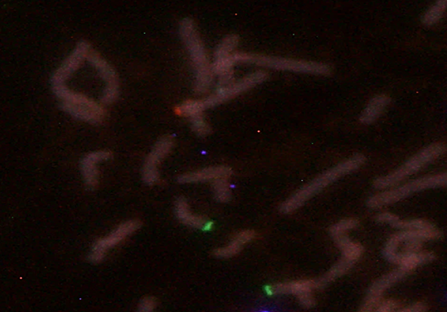
Se presenta el caso de una niña de 7 meses de vida derivada al Laboratorio de Citogenética del Instituto de Investigaciones en Ciencias de la Salud de la Universidad Nacional de Asunción en Paraguay (IICS - UNA), por presentar malformaciones diversas y alimentación nasogástrica, con sospecha de Síndrome de Wolf-Hirschhorn. La madre con 20 años y el padre con 26 años, sanos, no consanguíneos, refieren no haber tenido exposición a teratógenos. La niña es producto de un primer embarazo, nacida a término con 41 semanas de gestación, 2.320 g de peso e hipotonía generalizada. Al examen físico presentó microcefalia y foramen oval permeable, puente nasal chato, hipertelorismo, orejas dismórficas y de implantación baja, paladar hendido y retardo psicomotor (Figura 1 y 2). La presión pulmonar y el ritmo cardíaco normal con buena contractilidad. Para el diagnóstico citogenético, a la niña se le realizó el estudio cromosómico estándar (cariotipo) y la técnica de hibridación in situ fluorescente (FISH) para microdeleción del cromosoma 4p16. Para el FISH, se utilizó el protocolo de Sondas “Vysis” (Abbott Molecular Inc., USA), la sonda utilizada fue Vysis LSI IGH/FGFR3 DCDF para la región 4p16 spectrum Orange y 14q32 LSI IGH spectrum Green. Con las técnicas citogenéticas convencionales, se observó en el 100% de las células analizadas una translocación desequilibrada en el brazo corto del cromosoma 4, región 4p16.3, con una pieza extra de otro cromosoma de origen desconocido, resultado el Cariotipo: 46,XX,der(4)t(4p;?)(p16.3;?) (Figura 2 y 3). Y con el FISH, en el 100% de las células analizadas se observó una señal de espectro naranja para LSI FGFR3 (4p16.3) y dos señales de espectro verde para LSI IGH (14q32), la nomenclatura resultó: ishdel(4)(p16.3)(FGFR3-) (Figura 4) y 5). Ninguna anomalía fue observada en los cromosomas de los progenitores. Cariotipo de la madre: 46,XX Cariotipo del padre: 46,XY.
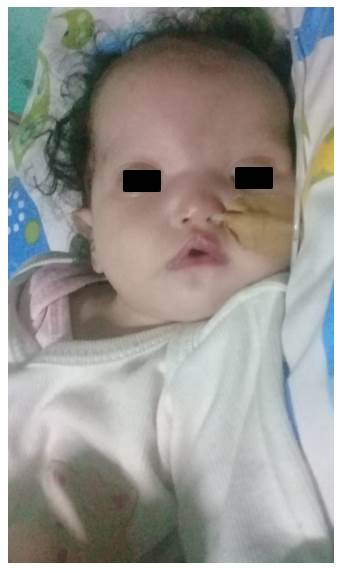
Figura 1 Se muestra la típica facies en casco de guerrero griego, caracterizado por puente nasal amplio que se extiende a la región frontal, se evidencia hipertelorismo.
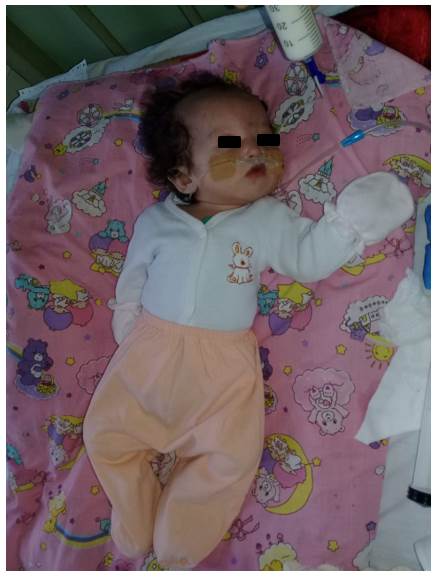

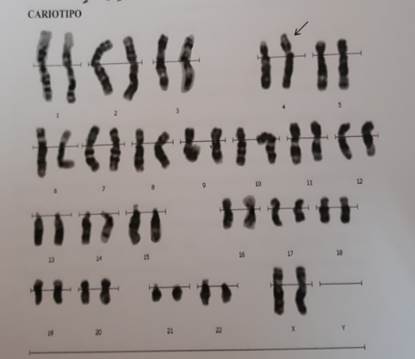
Figura 4 Kariograma donde se señala la translocación desequilibrada del brazo corto del cromosoma 4, región 4p16.3, con una pieza extra de otro cromosoma de origen desconocido. Cariotipo: 46,XX, der(4)t(4p;?)(p16.3;?).
DISCUSIÓN
El caso que se presenta como Síndrome de Wolf Hirschhorn, enfermedad que implica deleciones de tamaño variable en la región 4p16.3, está ubicada dentro del grupo de enfermedades raras6. En el cariotipo de la paciente se observó que el brazo corto de uno de los cromosomas 4 se encontraba aumentado de tamaño, suponiendo la presencia de una duplicación o translocación, en el análisis de las metafases con Bandas G, en primer lugar se descartó la duplicación porque con el FISH se detectó la microdeleción, quedando el cromosoma 4 alterado como portador de una translocación, pero no se ha podido identificar con que otro cromosoma se produjo la translocación. A fin de determinar el origen de la translocación, se procedió a realizar el cariotipo a los padres, en quienes sus cariotipos resultaron normales, por lo cual queda también establecido que se trata de un caso de novo, siendo una de las causas más frecuentes de los casos del síndrome de Wolf- Hirschhnorn7. Queda confirmada de esta manera la importancia de la realización de la técnica de Hibridación in situ fluorescente (FISH) en el caso de enfermedades raras, para una mayor confirmación, en el caso de una microdeleción8. Para estudios futuros la prueba de FISH de Painting o de Pintado cromosómico podría ser útil para determinar el origen de la translocación.