











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La hemofilia adquirida tipo A es una coagulopatía hemorrágica autoinmune, un raro trastorno de la hemostasia que produce autoanticuerpos que inactivan (inhiben) el F VIII, provocando sangrado en personas previamente sanas 1. Es una condición compleja que se sospecha en pocas ocasiones. Su primera presentación suele estar asociada con sangrado espontáneo de la piel, músculos, tejidos blandos y mucosas. Aunque su incidencia es baja, 1-1,5 casos por millón de habitantes por año, puede presentarse como una hemorragia súbita y severa 2,3. El sangrado severo y potencialmente mortal ocurre en 90% de los pacientes portadores 4.
Es más común en hombres entre las edades de 65 y 85 años. También puede ocurrir en 50% de las mujeres jóvenes entre 20 y 30 años durante el embarazo y el posparto o en asociación con otras condiciones: enfermedades autoinmunes, tumores sólidos, enfermedades de la piel y medicamentos. En 50% de los casos no se encuentra causa subyacente 5.
El plasma contiene F VIII como cofactor de la vía de coagulación intrínseca, por lo que la deficiencia de este provoca disfunción, reduce la generación de trombina y provoca hemorragias graves 6.
CASO CLÍNICO
Paciente de sexo masculino, de 60 años, de profesión mecánico, procedente de zona rural, presenta cuadro de 8 días de lesión violácea en cara interna del antebrazo izquierdo de inicio insidioso, no doloroso, pruriginoso, que con el transcurrir de los días se extiende abarcando todo el antebrazo con aparición de mismas lesiones en miembro superior derecho y región anterior del tórax. Al cuadro se agrega dificultad respiratoria de instalación progresiva, al principio a leves esfuerzos y luego a moderados esfuerzos, acompañado de debilidad generalizada por lo que decide acudir a nuestro servicio. Niega fiebre, picadura de insectos, traumatismos y consumo de fármacos en el último mes. Refiere cuadro similar anterior 2 años, pero no sabe referir diagnóstico ni tratamiento recibido. Paciente niega patologías de base.
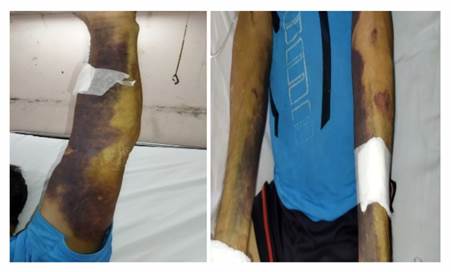
A su ingreso se constata palidez cutánea, piel con turgencia y elasticidad conservadas, sin edemas, hematoma que se extiende desde el hueco axilar hasta la muñeca en toda la cara antero interna del miembro superior izquierdo, no doloroso, sin aumento de la temperatura local (figura 1 y 2) sin estigmas de sangrado en mucosas.
En el hemograma se observa hemoglobina 5,6 g/L, hematocrito 16%, glóbulos blancos 6.020/mm3, neutrófilos 68%, linfocitos 24%, plaquetas 346.000/mm3. En frotis de sangre periférica: reticulocitos 3,7%, reticulocitos absolutos: 0,10%. Su TTPA era 159 seg. La orina simple no reporta hematuria y resto de la analítica sanguínea en rango.
Dentro del tamizaje de coagulopatía hemorrágica retornan resultados negativos de antígeno NS1, IgM para dengue, HBsAg, virus hepatitis C, VIH, SARS-CoV-2, ANA, anti-DNA, ENA y factor reumatoide. Anticuerpos anti-SSA/RO fue positivo a títulos bajos. El F VIII era 0,0%, inhibidor anti-F VIII 604,0 u/mL. Los marcadores tumorales fueron negativos. Ante test de guayaco positivo se realiza endoscopía digestiva alta que informa erosiones en antro sin sangrado activo. La colonoscopia mostró pólipo pediculado de gran tamaño de aspecto adenomatoso en colon transverso y pólipo sésil en número de 3, de aspecto adenomatoso, en colon transverso, pandiverticulosis colónica, hemorroides internas no congestivas. Ante el diagnóstico de hemofilia adquirida se decide iniciar ciclos de metilprednisolona con poca mejoría por lo que se inicia inmunoglobulina y administración de F VIII. Presentó evolución favorable posterior a la administración de ciclofosfamida a dosis bajas durante 6 meses con controles posteriores de TTPA de 40 segundos, F VIII80% e inhibidor no detectable, además de mejoría de lesiones señaladas en las figuras 1 y 2.
DISCUSIÓN
El caso presentado es poco frecuente, muchas veces no diagnosticado y en algunos con desenlace fatal. La hemofilia adquirida es una causa rara de sangrado en pacientes sin antecedentes personales o familiares de diátesis hemorrágica, con la incidencia más alta en mujeres jóvenes asociadas con embarazo o enfermedad autoinmune, y en hombres con una mediana de edad de 73 años. Según la literatura los sitios de sangrado más comunes son la piel, músculos, retroperitoneo y mucosas. La hemartrosis rara vez está presente en comparación con la hemofilia congénita 7,8.
Se diagnostica mediante pruebas de laboratorio. Un TTPA prolongado y un tiempo de protrombina normal indican un defecto en la vía de coagulación intrínseca y siempre deben investigarse. En primer lugar, debe probarse con plasma normal si se debe a la deficiencia de F VIII, el KPTT se verá alterado, mientras que en presencia de inhibidores del F VIII, el KPTT no se verá afectado 9.
El tratamiento de esta hemofilia tiene 2 aspectos: terapia hemostática y eliminación de inhibidores 10. En este paciente se basó en la administración de ciclos de metilprednisolona, en primera instancia con poca mejoría, por lo cual se decide la administración de inmunoglobulinas y F VIII con mejoría parcial por lo que se decidió la administración de ciclofosfamida.
Una vez establecido el diagnóstico, debemos iniciar tratamiento lo más rápido posible para evitar posibles complicaciones de dicha patología 11. En todo paciente con un TTPA prolongado asintomático debe estudiarse esta afección, antes que ocurran hemorragias graves. Después del diagnóstico, el tratamiento debe iniciarse lo antes posible, incluida la corrección de la hemostasia y la eliminación de los inhibidores. Una vez que se logra la erradicación y los parámetros hematológicos normales, las guías recomiendan un seguimiento mensual durante los próximos 6 meses y cada 3 meses a partir de entonces.

