











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) constituye una enfermedad compleja y variable en cuanto a su presentación morfológica, fisiopatológica y sintomatológica, así como en su pronóstico y sobrevida tanto en la vida adulta como en la pediátrica1. Se caracteriza por la hipertrofia del ventrículo izquierdo, aunque también podría presentarse en forma de hipertrofia biventricular o hipertrofia ventricular derecha aislada; que no se explica únicamente por condiciones de sobrecarga presóricas anormales, o no sean secundarias a otras patologías de base1,2.
La causa más importante de la MCH en adultos es debido principalmente a las mutaciones en los genes sarcoméricos, detectables en al menos una de las diez proteínas del sarcómero cardíaco. La MCH se produce por mutaciones en diferentes genes (heterogeneidad genética), aunque el modelo de transmisión es monogénico. Las mutaciones genéticas de las proteínas sarcoméricas suponen el 30-60% de todas las causas de MCH. La transmisión es autosómica dominante, y se han identificado entre 25 a 30 genes responsables3. Entre estas, las mutaciones en MYH7 y MYBPC3 están presentes en el 75% de los casos con variantes patogénicas identificadas. Mutaciones en otros genes sarcoméricos como MYL2, MYL3, TPM1, TNNT2, TNNI3, y ACTC1 solo son identificados en menos de 10% de los casos4-6. Por el contrario, en la edad pediátrica las causas no sarcoméricas, incluidos síndromes genéticos, trastornos metabólicos y mitocondriales, enfermedades de depósito y enfermedades neuromusculares degenerativas son las más frecuentes, y representan hasta el 35% de los niños con MCH2,6. La edad de presentación y la etiología impactan significativamente en el pronóstico. Por tanto, el diagnóstico etiológico es un paso fundamental en el complejo plan del manejo integral y la terapia específica en este grupo de pacientes.
La presentación clínica puede ser asintomática o presentar mareos, lipotimias, disnea, palpitaciones o dolor precordial referidos por pacientes de edad escolar o adolescentes, también puede presentarse como muerte súbita cardiaca (MSC) y ser diagnosticado posteriormente a través de una autopsia; en los lactantes pequeños los síntomas que no pueden ser referidos directamente, se detectan signos de insuficiencia cardiaca congestiva (ICC), como cansancio al mamar, llanto e irritabilidad incoercibles, dificultad respiratoria en reposo con quejidos, falla de medro, taquicardia, arritmia y hasta convulsión por bajo gasto cardiaco o MSC7.
A la auscultación podemos escuchar dos tipos de soplos sistólicos, el primero, si se trata de una MCH obstructiva, el soplo es de eyección en grado variable en mesocardio, foco aórtico y aórtico accesorio, el segundo más frecuente es el soplo sistólico de regurgitación a nivel de foco mitral que puede irradiar a todo el tórax incluida la espalda. El componente pulmonar del segundo ruido cardiaco puede estar aumentado con un click proto-sistólico de apertura en caso de existir una hipertensión pulmonar; otros signos de insuficiencia cardiaca o bajo gasto cardiaco como hepatomegalia y otros signos típicos de ICC pueden estar presentes pero no son patognomónicos o exclusivos de MCH, pues pueden ser secundarios a cualquier otra patología cardiaca, sin embargo, estos hallazgos sirven para estratificar el grado de compromiso o estadio en la evolución de la MCH.
Además de la anamnesis y el examen físico exhaustivo, la realización de estudios cardiológicos de imágenes no invasivas como el Electrocardiograma y Ecocardiografía Bidimensional y con Doppler Color, constituyen el primer paso para la confirmación diagnóstica ante la sospecha de la miocardiopatía7. La unanimidad de criterios en dicha evaluación, son universales y se basan principalmente, en realizar una completa evaluación ultrasonográfica secuencial y segmentar donde se descarta cualquier cardiopatía secundaria que sea causante de la MCH, a renglón seguido analizar los siguientes datos morfológicos y fisiológicos-funcionales a ser definidos, y que deberían ser incluidos en la descripción y conclusión de un Ecocardiograma Doppler Color con diagnóstico de MCH8:
Dichos criterios morfológicos y fisiológicos de MCH son:
1- Patrón y severidad del engrosamiento miocárdico, aunque visualmente la hipertrofia de las paredes del ventrículo izquierdo pudieran ser notorias en la modalidad bidimensional, es necesaria la medición en diástole del grosor de las paredes ventriculares. La Sociedad Europea de Cardiología (ESC) define como hipertrofia en los pacientes adultos cuando el grosor en diástole de la pared ventricular es ≥15 mm; en los pacientes pediátricos cuando son ≥2 desviaciones estándar con respecto a la media del grosor para su edad. En el diagnóstico de familiares de primer grado de pacientes con MCH, es necesario un grosor del VI ≥13 mm6,8. En cuanto a la morfología de la hipertrofia es importante definir el tipo morfológico de MCH, si es septal basal, medio septal o septal difuso, apical o concéntrica global o difusa. Figura 1A-B.
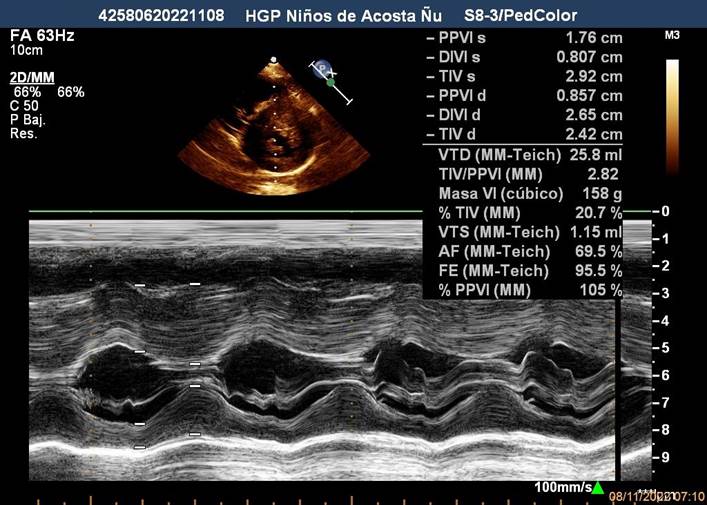
Figura 1A Imagen Ecocardiográfica Transtorácica Bidimensional en Modo M, donde se observa el aumento importante del grosor de la pared ventricular izquierda a predominio septal, disminución de la cavidad ventricular en diástole, aumento de la fracción de eyección y de acortamiento, los cuales representan criterios diagnósticos de MCH en Modo M.
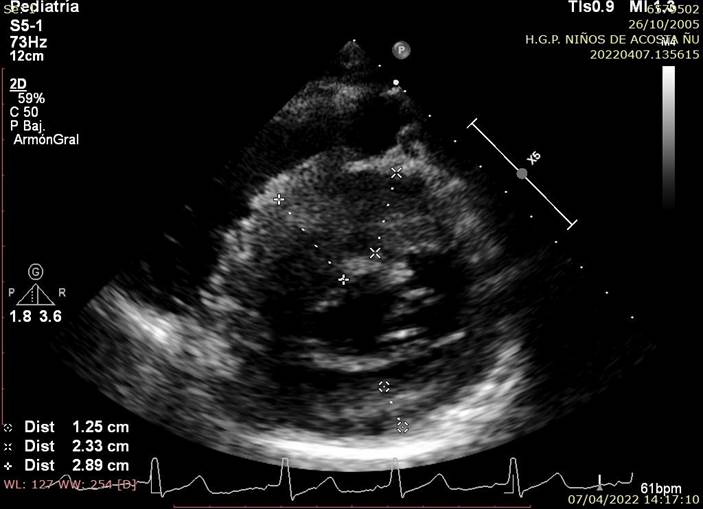
Figura 1B Imagen Ecocardiográfica Transtorácica Bidimensional en vista Paraesternal eje corto, donde se visualiza la gran hipertrofia concéntrica ventricular con predominio a nivel septal midiendo 28.9mm de grosor.
2- Además de definir el patrón y severidad del engrosamiento miocárdico, es importante determinar la presencia y naturaleza de la obstrucción de salida al flujo sanguíneo ventricular izquierdo y/o derecho, dicha obstrucción es consecuencia directa del grado de hipertrofia de la MCH.
3- Es necesario buscar la presencia del movimiento sistólico anterior de la válvula mitral (SAM) y cuantificar la severidad de la regurgitación resultante.
4- Medir el tamaño de la aurícula izquierda.
5- Definir el patrón de llenado diastólico del ventrículo izquierdo, y por último,
6- Determinar y cuantificar la presencia o no de hipertensión pulmonar.
La vista apical de cuatro cámaras y la de paraesternal eje largo o longitudinal, demuestran con mayor claridad los hallazgos de MCH. En los casos obstructivos se reconoce un engrosamiento prominente del septum basal, distorsión de las estructuras de soporte mitral, reducción del diámetro del tracto de salida del flujo asociado con un movimiento sistólico de la válvula mitral anterior. Figura 1 C.
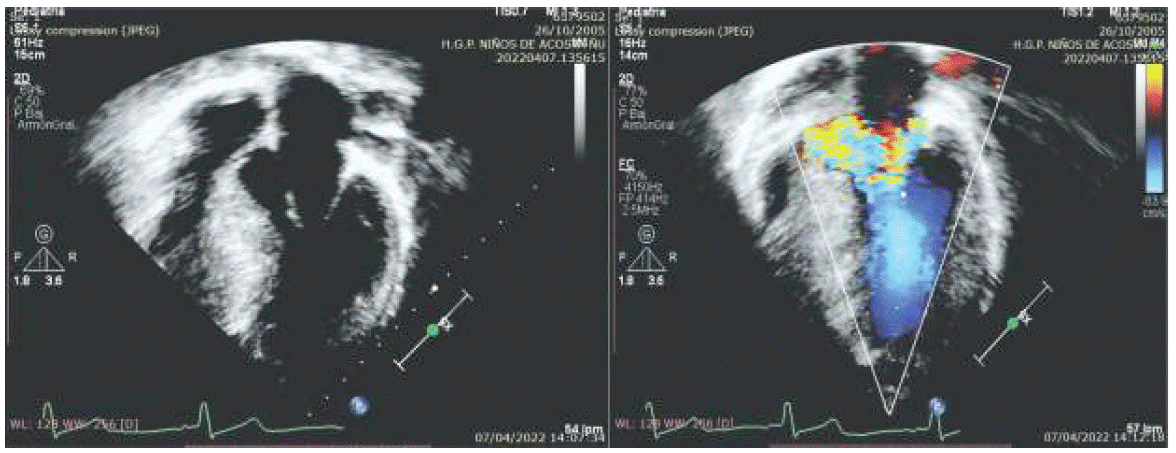
Figura 1C Imagen Ecocardiográfica Transtorácica Bidimensional en vista apical 4 cámaras y con Doppler Color donde se visualiza un engrosamiento prominente del septum basal, distorsión de las estructuras de soporte mitral y reducción del diámetro del tracto de salida del ventrículo izquierdo.
El Doppler de flujo en color muestra y localiza la turbulencia asociada con la obstrucción dinámica de salida del flujo. El Doppler de onda continua permite la cuantificación de las velocidades de flujo obstructivo, además muestra un perfil característico “en forma de Daga”. Figura 2 A-B.
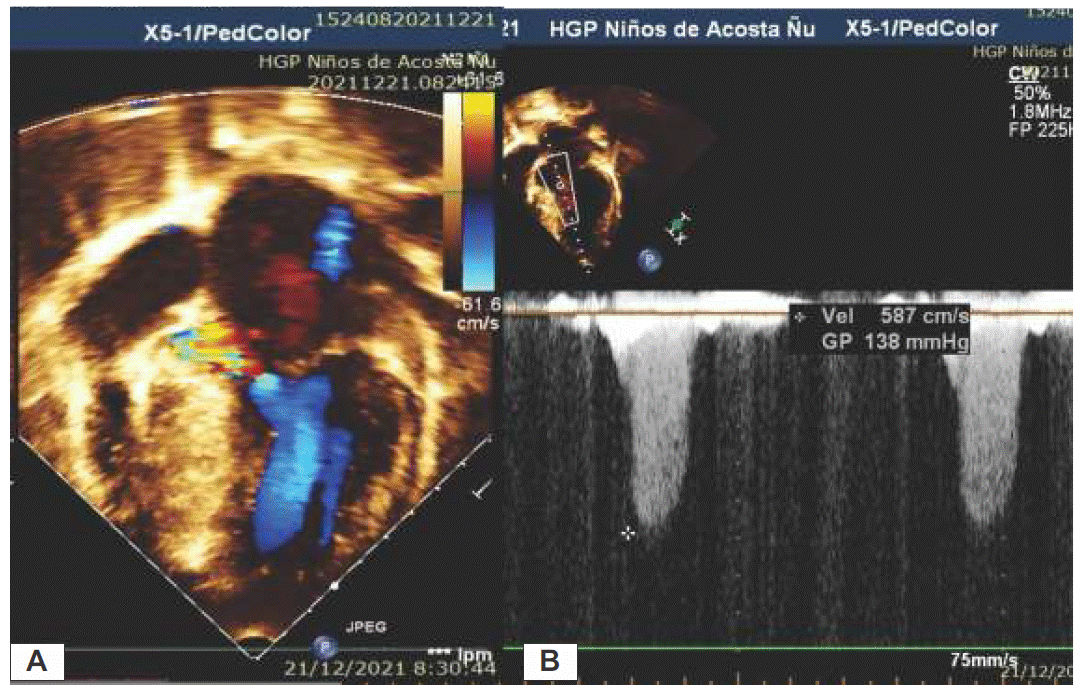
Figura 2A Imagen Ecocardiográfica Transtorácica Bidimensional en vista apical 4 cámaras con Doppler Color donde se demuestra la obstrucción dinámica del tracto de salida del ventrículo izquierdo. B. Imagen del Doppler Continuo con velocidad aumentada debido a la obstrucción en el tracto de salida generando un gradiente de 138mmHg.
En la valoración de la disfunción diastólica se analiza la prolongación del tiempo de relajación isovolémica y la reducción de la velocidad de llenado temprano transmitral. En la medida que la fibrosis miocárdica aumenta, el patrón de Doppler se aproxima a un patrón de tipo restrictivo. También se evalúa la disminución de las velocidades de Doppler de tejido anular mitral diastólico tempranas. La comparación de la velocidad de llenado diastólico transmitral temprano mediante Doppler de onda pulsada con la velocidad Doppler de tejido anular temprana (proporción E/E´) cuyos valores más grandes se asocian con una enfermedad más avanzada8.
El Electrocardiograma (ECG), puede presentar alteraciones que podrían ser la única manifestación de la enfermedad en una etapa inicial o precoz. Aproximadamente en el 90% de los pacientes con diagnóstico de MCH presentan alteraciones en el ECG, aunque ninguno sea patognomónico o exclusivo. Los hallazgos más frecuentes son los trastornos de la repolarización que están presentes en alrededor del 50% de los pacientes con hipertrofia del ventrículo izquierdo, de los cuales se pueden encontrar cambios en el punto J, descenso del segmento ST y onda T negativas en derivaciones izquierdas. La presencia de onda T positiva en aVR está asociada a eventos arrítmicos mayores como muerte súbita cardíaca, taquicardia ventricular sostenida y/o fibrilación ventricular. Otros hallazgos pueden ser signos de dilatación auricular derecho y/o izquierdo; trastornos en la conducción intraventricular presentándose como fragmentación del QRS, ya sea por la presencia de onda R mellada o una muesca en la onda S, constituyéndose así un marcador de fibrosis en el miocardio. La prolongación del intervalo QT está presente hasta en 60% de los pacientes. Algunas MCH que se encuentran relacionadas a enfermedades de depósito o a síndromes genéticos pueden presentar vías accesorias de preexcitación como el síndrome de Wolff Parkinson White6,7,9. Figuras 3A-B-C.
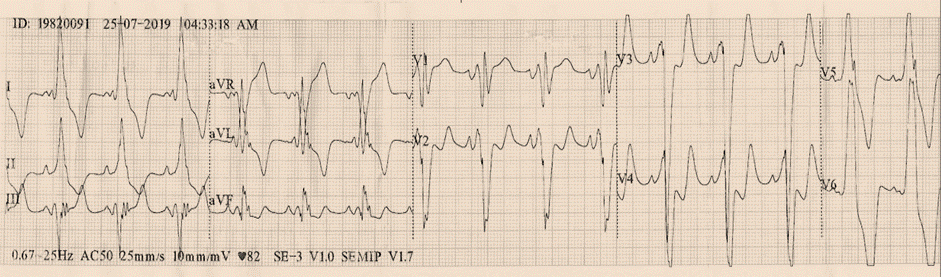
Figura 3A Trazado electrocardiográfico. Presencia de onda R adicional y muesca en la onda S, que refleja la demora de la conducción intraventricular. Trastorno de la repolarización con infradesnivel del segmento ST y onda T negativas en derivaciones izquierdas. Onda T positiva en aVR, hallazgo asociado a eventos arrítmicos mayores.

Figura 3B Trazado electrocardiográfico en paciente con MCH asociado a Enfermedades de depósito. Presencia de preexcitación, Síndrome de Wolff-Parkinson-White.
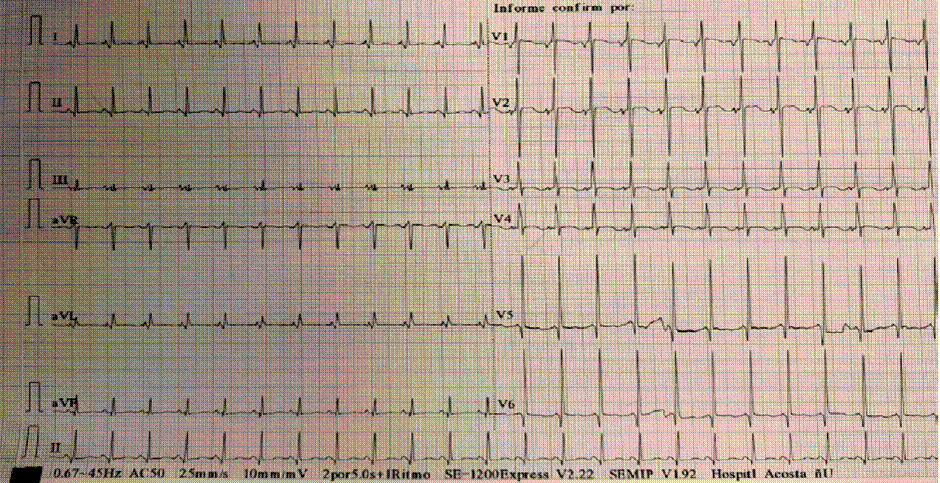
Figura 3C Trazado electrocardiogáfico en paciente con MCH. Trastorno de la repolarización ventricular, con cambios en la morfología de la Onda T. Prolongación del intervalo QTc.
Otro de los métodos de diagnósticos por imágenes que actualmente cumple un rol muy importante es la resonancia magnética cardiaca (RMC), en especial para la diferenciación de la MCH de la hipertrofia cardíaca en los atletas, enfermedad hipertensiva u otras miocardiopatías. La RMC es el método de elección para la evaluación de la fibrosis miocárdica y su extensión con el empleo de sustancias de contraste. En varios estudios describen una relación directa y continua entre la extensión de fibrosis y el riesgo de los eventos de MSC1,6,9,10).
Las imágenes de RMC aportan una clara ventaja para mediciones del espesor del miocardio, para cuantificar la masa del ventrículo izquierdo, tamaño del ventrículo derecho; determinar la función sistólica e identificar áreas focales de hipertrofia del ventrículo izquierdo, principalmente si la hipertrofia se limita a determinadas regiones de la pared ventricular, como la pared posterior, anterolateral, tabique y ápice; determinar anormalidades de la válvula mitral y aparato subvalvular, características específicas de la anatomía del tracto de salida del ventrículo izquierdo, además de anomalías adicionales como la presencia de aneurismas apicales del VI que no son muy claros en vistas ecocardiográficas. La mayor sensibilidad en la utilización de imágenes de RMC se atribuye a su alta resolución espacial, además de que dichas imágenes no son obstaculizadas por ventanas acústicas deficientes causadas por obesidad, parénquima pulmonar, deformidades torácicas, entre otras1,6,11. Figura 4A-B-C-D-E.
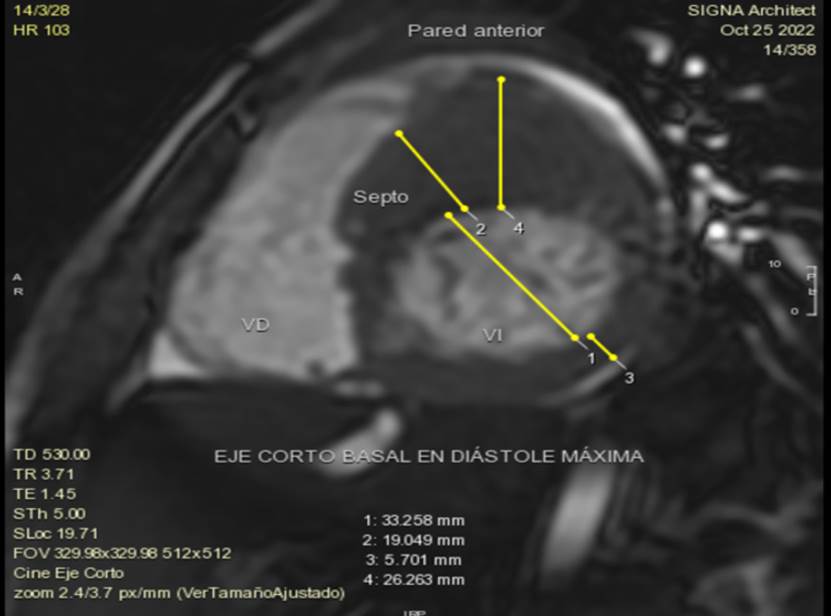
Figura 4A Imagen de Cine Resonancia Magnética Cardiaca de corte eje corto basal, exhibiendo hipertrofia parietal importante de la pared anterior y segmento anteroseptal.
Figura 4B Imagen de Cine Resonancia Magnética Cardiaca en 4 cámaras. Aumento del espesor parietal izquierdo con predominio septal septal.
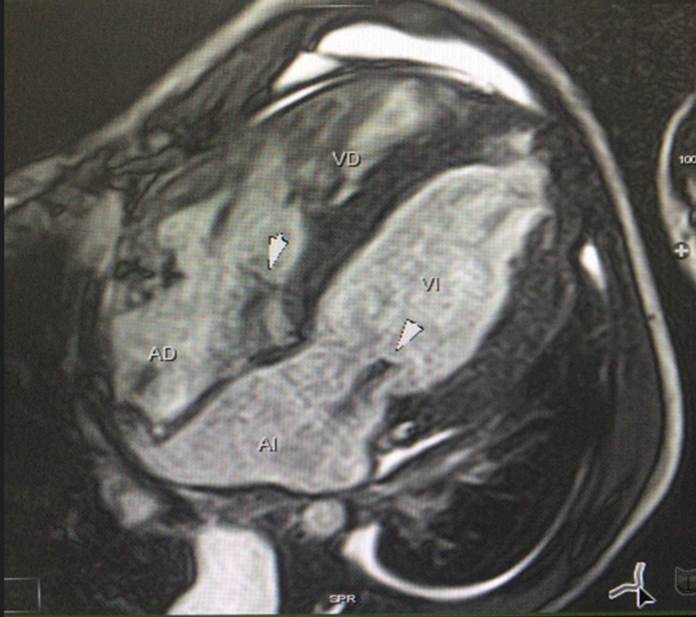
Figura 4C Imagen de Cine Resonancia Magnética Cardiaca en 4 cámaras. Se observa hipertrofia difusa del ventrículo izquierdo, además de jet de regurgitación tricúspide y mitral.
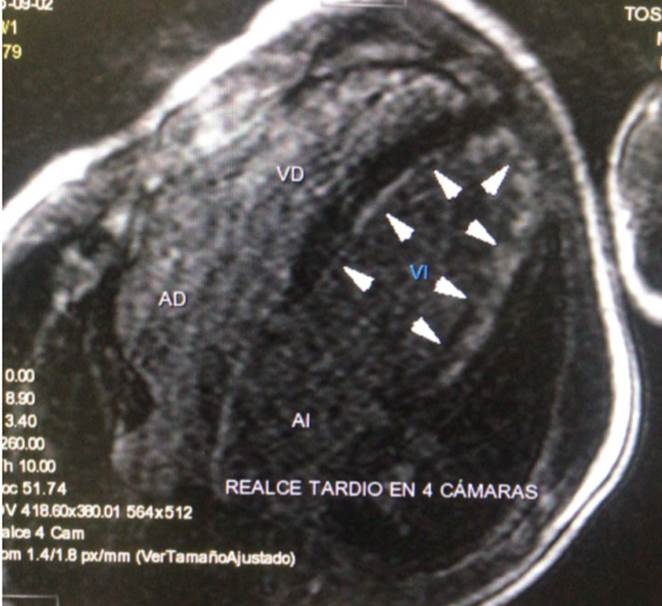
Figura 4D Imagen de Cine Resonancia Magnética Cardiaca en 4 cámaras, con realce tardío. Se observa realce tardío subendocárdico circunferencial difuso.

Figura 4E Imagen de Cine Resonancia Magnética Cardiaca en eje corto. Se observa realce tardío subendocárdico circunferencial difuso con afectación transmural en el segmento anteroseptal basal (A) y medial (B).
En cuanto a la aplicación de pruebas genéticas para un diagnóstico específico, existen en la actualidad la secuenciación masiva del ácido desoxirribonucleico, que permite detectar nuevos genes responsables de la enfermedad, aunque tienen el inconveniente de poder detectar algunas variantes con significado incierto3,4. Sin embargo, ante la presencia de sospechas clínicas convincentes en pacientes con MCH, existen paneles genéticos integrales para miocardiopatías que proporcionan un análisis completo de los genes asociados con las miocardiopatías más conocidas hasta la fecha. Las recomendaciones de los genes a analizar en las pruebas genéticas, debido a su gran heterogeneidad de más de 1.500 mutaciones descritas, se limita a 8 genes del sarcómero (MYH7, MYBPC3, TNNT2, TPM1, MYL2, MYL3, TNNI3 y ACTC1) y 3 metabólicos (GLA, LAMP2, PRKAG2), los cuales presenta una etiología bien establecida3).
Cómo pueden coexistir superposición clínica entre diferentes etiologías de miocardiopatías, las pruebas genéticas integrales permiten una evaluación más eficiente de múltiples condiciones como: Enfermedades de depósitos como Pompe, Fabri, Danon, las Mucopolisacaridosis, entre otras como Ventrículo Izquierdo no compactado, Trastornos del Espectro Noonan, Displasia arritmogénica del ventrículo derecho, etc., por lo que es de suma importancia especificar cuáles son las sospechas diagnósticas más fuertes a ser confirmadas.
MATERIALES Y MÉTODOS
Estudio observacional, descriptivo, de corte transversal, de una serie de casos de pacientes con diagnóstico de MCH de diferentes etiologías, atendidos en el Servicio de Cardiología Pediátrica del Hospital General Pediátrico “Niños de Acosta Ñu”, en el periodo comprendido de enero de 2020 a diciembre de 2022.
RESULTADOS
Se han analizado 12 pacientes atendidos en el Servicio de Cardiología Pediátrica del Hospital General Pediátrico “Niños de Acosta Ñu”, en el periodo comprendido de enero de 2020 a diciembre de 2022, con edades que varían desde 6 meses hasta 16 años, con diagnóstico de MCH con presentación clínica variable y de distintas etiologías. A continuación, se detallan los casos en la Tabla 1.
Tabla 1 Datos clínicos, diagnósticos por imágenes y genéticos.
| Edad | Sexo | Motivo de consulta | Ecocardiografía | Estudio Genético | Enfermedad | Evolución |
| 15 años | M | Insuficiencia cardiaca congestiva | Miocardiopatía hipertrófica, no obstructiva, con disfunción sistodiastólica biventricular severa | Gen LAMP2 | Enfermedad de Danon | Ingresa a lista de trasplante cardiaco, presenta muerte súbita cardiaca en espera. |
| 9 meses | M | Soplo cardiaco, facie dismórfica con cuello alado, hipotrofia e hipotonía muscular. | MCH concéntrica biventricular no obstructiva y estenosis valvular pulmonar leve (gradiente 35 mmHg). | Gen BRAF, exón 11, c.1391G>T/p.Gly464Val en heterocigosis | Síndrome Cardio-Facio-Cutáneo | Clase funcional II/IV (Clasificación modificada de Ross), Medicación: Propranolol vía oral. |
| 10 años | M | Talla baja, retraso mental, facie peculiar. | MCH moderada a severa, no obstructiva. | Panel para enfermedades de depósito | Mucopolisacari-dosis Tipo I. | Clase Funcional III/IV (NYHA) Medicación: Carvedilol, vía oral. En tratamiento con terapia de reemplazo enzimático. |
| 6 años | M | Soplo cardiaco, electrocardiograma con patrón de Wolff-Parkinson-White (WPW). | MCH concéntrica no obstructiva biventricular | Estudio genético para 162 clases de miocardiopatías en asociación con WPW, resultó negativo en 2 estudios consecutivos. | Clase funcional I/IV (NYHA). En seguimiento clínico cardiológico, aún sin medicación. | |
| 8 años | M | Presentó taquicardia paroxística supraventricular a los 6 años de edad, electrocardiograma y Holter de 24 horas patrón de WPW. | MCH septal asimétrica con predominio basal, y elevación de la presión sistólica pulmonar | Gen MYH7.c(w).p.(Gly7116Arg).(wt). | MCH tipo 7 | Clase funcional I/IV (NYHA). En seguimiento clínico cardiológico, Amiodarona vía oral |
| 6 años | F | Insuficiencia cardiaca congestiva global grado IV/IV (NYHA) | MCH restrictiva en fase dilatada, con disfunción sistodiastólica severa e hipertensión pulmonar severa (62mmHg). | Gen TNNI3 en heterogicosis | Miocardiopatía familiar restrictiva tipo 1 y MCH tipo 7. | Recibió asistencia biventricular (Berlin Heart, BH) por 5 días. Trasplante cardiaco con éxito en julio 2022. |
| 16 años | F | Soplo cardiaco. Múltiples léntigos en el cuerpo, frente amplia, hipertelorismo, inclinación palpebral antimongoloide. | MCH con severa obstrucción del tracto de salida del ventrículo izquierdo, a predominio septal. | Síndrome de Noonan con Múltiples Léntigos | Clase funcional II/IV (NYHA). Medicación: Propranolol. Portadora de cardiodesfibrilador. | |
| 6 años | F | Cansancio a grandes esfuerzos, en consulta se ausculta soplo cardiaco. | MCH no obstructiva. | Gen RAF1, c.782C (Pro261Leu) | Síndrome de Noonan. | Clase funcional II/IV (NYHA). Medicación: Carvedilol. |
| 5 meses | F | Ingresa a UCIP en shock cardiogénico | MCH no obstructiva con disfunción severa | Panel para enfermedades de depósito | Enfermedad de Pompe | Óbito durante la internación en UCIP antes de recibir la terapia de reemplazo enzimático. |
| 14 años | F | Cansancio a moderados esfuerzos, marcha tambaleante, pérdida de coordinación de la motricidad fina. | MCH moderada no obstructiva | Panel genético. | Enfermedad degenerativa de Ataxia de Friedreich. | En seguimiento ambulatorio por cardiología. Medicación: Propranolol. |
| 1 año | M | Dificultad respiratoria | MCH no obstructiva en fase dilatada con disfunción sistodiastólica biventricular severa | TAZ;Exon 3;c.[80C>T]/p.[Arg94Cys] | Síndrome de Barth | Ingresa a lista de Trasplante cardiaco. Óbito durante la internación en UCIC conectado a ECMO. |
| 6 años | F | Dificultad respiratoria y cansancio a mínimos esfuerzos. | MCH no obstructiva en fase dilatada con disfunción sistodiastólica biventricular severa. | PKP2;Intron 13;c.[2578-2A>C]; [2578-2A>C] | Miocardiopatía arritmogénica del ventrículo derecho. | Recibió asistencia biventricular (BH) durante 3 meses. Trasplante cardiaco con éxito en octubre 2022. |
MCH: miocardiopatía hipertrófica. NYHA: New York Heart Association. WPW: Wolff-Parkinson-White. UCIP: Unidad de Cuidados Intensivos Pediátricos. UCIC: Unidad de Cuidados Intensivos Cardiovasculares.
DISCUSIÓN
Las características clínicas y morfológicas de la MCH en la edad pediátrica son similares a las del paciente adulto, sin embargo, hay diferencias cuando nos referimos al pronóstico. La mortalidad de MCH en lactantes a los 2 años del diagnóstico es alrededor del 30%, mientras que los que son diagnosticados en edades más tardías como en los escolares o adolescentes tienen una mayor sobrevida9. La sobrevida es mayor cuando se hace el diagnóstico a partir del primer año de vida, pudiendo llegar cerca del 97% a los 5 años y alrededor del 94% a los 10 años de realizado el diagnóstico2,9; aunque debemos aclarar que esta evolución se encuentra directamente relacionada al origen genético de cada grupo específico de MCH. El primer pico de mortalidad es durante el primer año de vida que está causada principalmente por síndromes malformativos o por errores hereditarios del metabolismo y el segundo pico se presenta alrededor de los 8 y 12 años relacionadas con mutaciones en genes sarcoméricos2,9, dichos datos de la literatura se correlacionan con nuestra serie de casos. Las principales causas de muerte son la MSC secundaria a arritmias, insuficiencia cardíaca y tromboembolismo2,7,9,10,11.
Los pacientes con MCH asociados a Rasopatías (Síndrome de Noonan, Síndrome Cardio-Facio-Cutáneo, Síndrome de Noonan con Múltiples Léntigos), presentan similares característica clínicas y evolutivas, presentan igual tasa de mortalidad anual que aquellos pacientes que no presentan MCH. La tasa de mortalidad es alrededor del 9%, con una edad media de sobrevida de 60 años2,12.
Las MCH familiares son las que tienen mayor morbimortalidad, con una forma de presentación más precoz, peor pronóstico debido a la elevada incidencia de MSC, importante fibrosis y frecuente necesidad de cardiodesfibrilador implantable o terapia de resincronización1,6,7,9.
Los casos que se asocian con Enfermedades de depósito como la Enfermedad de Danon son los de peor pronóstico como lo hemos encontrado en el caso presentado. El daño cardíaco global es causado por el deterioro de la función de la proteína LAMP2 que conduce a la acumulación de material autofágico y glucógeno, siendo la insuficiencia cardiaca y las arritmias las principales causas de la elevada morbilidad y mortalidad2,13. Otras enfermedades de Depósito como la Enfermedad de Pompe en la forma de presentación infantil tiene una elevada mortalidad, que coincide con nuestro caso de 5 meses de edad, y que es debido a la temprana afectación cardiovascular, con evolución a insuficiencia cardíaca refractaria y muerte en el primer año de vida2,14. En los casos de Mucopolisacaridosis presentan una afectación variable en el sistema cardiovascular, afectando al miocardio, aparato valvular, tejido de conducción y/o vascular. El pronóstico varía según el tipo de Mucopolisacaridosis, siendo el tipo I el fenotipo más grave. La causa cardiovascular de muerte se debe principalmente a la insuficiencia cardíaca avanzada, mientras que la MSC o la muerte por isquemia coronaria son raras2,15. La finalidad de detectar precozmente estas enfermedades es el inicio oportuno de la terapia de reemplazo enzimático que mantiene la clase funcional, retarda o reduce el compromiso infiltrativo miocárdico y por lo tanto extiende notablemente la sobrevida.
En enfermedades degenerativas con afectación cardiovascular como la Ataxia de Friedreich, al igual que el caso de nuestro estudio, generalmente se presenta con una hipertrofia del ventrículo izquierdo de manera inexplicable, manifestándose durante la adolescencia y está presente en más de la mitad de los pacientes; en raros casos, la enfermedad progresa a dilatación y a progresiva disfunción ventricular lo que puede provocar insuficiencia cardiaca y muerte2,16.
La miocardiopatía en el Síndrome de Barth se presenta aproximadamente en el 90% de los varones, sin embargo, la manifestación y gravedad puede variar en cada individuo afectado. Se describen diversos fenotipos cardiomiopáticos que incluyen la no compactación del ventrículo izquierdo y la miocardiopatía dilatada, solos o en conjunto con la fibroelastosis endocárdica, MCH, y una forma apical de MCH, entre otros. La insuficiencia cardiaca constituye una de las principales causas de temprana mortalidad, lo que coincide con nuestra serie de casos del paciente masculino de un año de edad, seguida por las arritmias que son frecuentes y pueden ser potencialmente mortales2,17,18.
La miocardiopatía arritmogénica del ventrículo derecho presenta una incidencia de 1,01 cada 100.000 niños al año entre los 15 y 19 años, siendo muy rara antes de los 10 años de edad, como lo hemos encontrado en esta serie de casos. En la actualidad el espectro de manifestaciones se ha ampliado, reconociendo de esa manera manifestaciones difusas en el VD, afectación única del ventrículo izquierdo y alteración biventricular en fase dilatada19. Si bien es de infrecuente presentación en la edad pediátrica, es importante reconocer esta entidad por la posibilidad de mejorar el pronóstico de pacientes que se podrían beneficiar con el implante de un cardiodesfibrilador, debido a la alta incidencia de arritmias ventriculares y muerte súbita. La insuficiencia cardíaca es una manifestación tardía de esta entidad19,20.
El único tratamiento que ha demostrado ser eficaz en la prevención de la MSC es el implante precoz de un cardiodesfibrilador en pacientes seleccionados1,5,6,7,9-11,18,20. Las recomendaciones de tratamiento farmacológico persiguen los objetivos de optimizar la capacidad funcional reduciendo síntomas, disminuir riesgo de MSC y mejorar la supervivencia1,2,6,7,9,10. La realización de estudios imagenológicos acompañado de paneles genéticos es primordial para el diagnóstico certero y de esta manera determinar la estrategia apropiada con la aplicación de un tratamiento precoz, oportuno y efectivo. Se destaca la importancia de la realización de paneles genéticos en pacientes con MCH, aunque actualmente en nuestro país no se realizan dichos estudios por lo que las muestras son enviadas al extranjero para su debido procesamiento.
El tratamiento farmacológico tiene como principal objetivo el alivio de los síntomas, porque no hay datos convincentes que indiquen que el tratamiento farmacológico pueda alterar la historia natural de la MCH6. El éxito de la medicación está determinado por la respuesta de los síntomas del paciente6,7,9,10. La mayoría de los pacientes en nuestro estudio reciben beta-bloqueantes como el Propranolol y el Carvedilol, que son considerados de primera línea descritos en la literatura. Los bloqueadores de canales de calcio, como el verapamilo o diltiazem son alternativas razonables en pacientes mayores de un año de edad6,7,9, sin embargo no contamos con experiencia con dicha medicación en la serie de casos presentada. En pacientes con MCH que presentan arritmias ventriculares frecuentes, la terapia de primera línea son los betabloqueadores. Sin embargo, si la taquicardia ventricular es recurrente, se pueden agregar otros agentes antiarrítmicos como la amiodarona, pese a que presente efectos secundarios, no presenta efecto sobre la supervivencia global6.
Para aquellos pacientes que no responden a los beta-bloqueadores o a los bloqueadores de los canales de calcio no dihidropiridínicos y cuyos síntomas deterioran la calidad de vida, podrían beneficiarse de otras terapias más avanzadas como la disopiramida, que no se encuentra disponible en nuestro país, y la terapia de reducción septal (TRS)1,2,6,7,9.
Los criterios generales para la TRS son: a) Clínicos: como dolor torácico o disnea severa (clase funcional III o IV de la NYHA), u otros síntomas de esfuerzo (síncope, casi síncope), cuando se atribuye a obstrucción de vía de salida del ventrículo izquierdo. b) Hemodinámico: presencia de gradiente dinámico pico de 50 mmHg en el tracto de salida del ventrículo izquierdo en reposo o provocados con cambios fisiológicos, que se encuentra asociado con hipertrofia septal y movimiento sistólico anterior (SAM) de válvula mitral. c) Anatómico: Grosor de la pared septal anterior suficiente para realizar el procedimiento de manera segura y efectiva6.
La vía clásica para el abordaje de la miectomía septal transaórtica se encuentra limitado en lactantes y niños pequeños, debido al pequeño tamaño del anillo aórtico, en dichos pacientes el procedimiento de Konno modificado proporciona resultados a largo plazo de manera igualmente satisfactorios6.
La hipotensión aguda representa una urgencia médica en los pacientes con MCH obstructiva. El objetivo del tratamiento consiste en maximizar la precarga y la poscarga, evitando el aumento de la contractilidad o frecuencia cardíaca. Los fármacos vasoconstrictores endovenosos como la fenilefrina pueden revertir esta delicada situación. Los betabloqueadores también pueden ser útiles, siempre en combinación con un vasoconstrictor ya que disminuye la contractilidad y mejora la precarga al prolongar el período de llenado diastólico6.
La estratificación del riesgo para MSC en niños con MCH requiere del análisis de varios factores de riesgo. La presencia de taquicardia ventricular no sostenida, el síncope inexplicable, el tamaño del grosor de la pared del ventrículo izquierdo y las puntuaciones Z del diámetro auricular izquierdo tienen una relación parecida con el riesgo MSC en pacientes pediátrico que en adultos. Sin embargo, en relación con la edad, los antecedentes familiares de MSC y el gradiente en el tracto de salida del ventrículo izquierdo difieren en comparación con los adultos. Se han desarrollado modelos para predecir el riesgo MSC, pero en la práctica clínica aún no se están utilizando ampliamente. Para la prevención primaria se recomienda considerar el uso de cardiodesfibrilador implantable en niños con MCH con la presencia de uno de estos factores de riesgo principales6, como se ha realizado en uno de los pacientes presentados.
Se debe considerar la evaluación de trasplante cardiaco según las guías vigentes en pacientes con taquiarritmias ventriculares potencialmente mortales, mal toleradas, recurrentes, refractarias a terapia con fármacos antiarrítmicos en dosis máximas2,6,7. Hemos optado por el ingreso a lista de trasplante cardiaco a tres de los pacientes estudiados, debido al avanzado deterioro de la clase funcional, de los cuales un paciente presentó muerte súbita cardiaca y los otros dos recibieron asistencia biventricular (Berlín Heart), con posterior trasplante cardiaco exitoso.
Con relación a la realización de ejercicio recreativo de leve a moderada intensidad se considera beneficioso para mejorar el funcionamiento físico, el estado cardiorrespiratorio y la calidad de vida. Los niños en general pueden participar en educación física en la escuela6.
CONCLUSIÓN
La MCH es una patología compleja y variable en cuanto a su presentación morfológica, fisiopatológica y sintomatológica, así como en su pronóstico y sobrevida. La MCH de causas no sarcoméricas como los síndromes malformativos y errores innatos del metabolismo son las que tienen peor pronóstico y sobrevida. Ante la sospecha diagnóstica de esta entidad es de crucial importancia la realización de un examen físico exhaustivo, con el apoyo de los métodos de imágenes cardiovasculares y con la realización de pruebas genéticas, dichos pacientes pueden beneficiarse al establecer o confirmar el diagnóstico, aclarar los riesgos, iniciar tempranamente la terapia específica, realizar un seguimiento y asesoramiento apropiado, para así retrasar el compromiso cardiovascular y mejorar la sobrevida. Las personas asintomáticas dentro de una familia con una variante patogénica conocida también pueden beneficiarse al evitar actividades y medicamentos que pueden desencadenar síntomas.

