











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome de interrupción del tallo hipofisario es una alteración congénita rara de la glándula hipófisis, que se asocia a deficiencia aislada de la hormona del crecimiento u otras hormonas secretadas en la hipófisis anterior1. La prevalencia exacta es incierta, un informe del año 2013 reportó 0.5 casos por cada millón de nacimientos2. Radiológicamente, el síndrome de interrupción del tallo hipofisario se caracteriza por la tríada clásica de: tallo hipofisario interrumpido o ausente, hipófisis posterior ectópica e hipoplasia o aplasia de la hipófisis anterior3. La manifestación clínica del síndrome depende de la edad al momento de establecer el diagnóstico. En los recién nacidos aparece como hipoglucemia, ictericia, criptorquidia y micropene. En los adolescentes y adultos se caracteriza, principalmente, por talla baja, retardo en los caracteres sexuales, epilepsia, retraso intelectual o ambos4. La causa del síndrome de interrupción del tallo hipofisario aún se desconoce, pero se han propuesto 2 teorías: la primera son lesiones perinatales y la segunda una organogénesis defectuosa, provocadas por factores genéticos o ambientales durante el embarazo5. Un diagnóstico tardío de la enfermedad determina una significativa morbimortalidad, lo que demuestra la importancia de identificar signos y síntomas clínicos presentes en etapas tempranas de la vida, especialmente en el período neonatal. La instauración de un tratamiento adecuado permitiría evitar el deterioro de la función cognitiva, los efectos deletéreos derivados de las múltiples deficiencias de hormonas hipofisarias y el abordaje apropiado de los defectos congénitos asociados6. A continuación presentamos el caso de una paciente con interrupción de tallo hipofisario diagnosticado en la edad adulta.
REPORTE DE UN CASO
Paciente de sexo femenino, 24 años de edad, acude al consultorio de Endocrinología por amenorrea. Como antecedentes personales de valor, fue nacida por parto eutócico, no traumático, a las 38 semanas de gestación, con talla al nacer de 48cm; tuvo adecuado desarrollo psicomotor hasta la actualidad. Como dato de valos, la talla diana familiar es de 168 ± 5 cm.
Al interrogatorio refiere antecedente de terapia de reemplazo hormonal con valerato de estradiol y norgestrel, además de hormona de crecimiento desde los 15 hasta los 19 años, que luego abandona, sin saber detallar el motivo y tampoco cuenta con expediente clínico. Niega trastornos alimentarios, consumo de medicación o actividad física extenuante.
Al examen físico se constatan caracteres sexuales secundarios poco desarrollados, talla baja e índice de masa corporal normal.
Se solicita perfil hormonal (Tabla 1) donde se constata hipogonadismo hipogonadotrófico y deficiencia de hormona de crecimiento. Se solicita ecografía ginecológica donde se observa útero y ovarios de tamaño disminuidos, con ecoestructura homogénea.
Tabla 1. Perfil hormonal
| Parámetro | Valor |
|---|---|
| LH | 0,5 IU/L (0,7-5,6) |
| FSH | 2,2 IU/L (2,5-10) |
| Estradiol | 16,3 pg/ml (19,5-144,2) |
| IGF-1 | 35 ng/ml (109-284) |
| PRL | 2,5 ng/ml |
| Testosterona | 13,4 |
| TSH | 2,2 mIU/L |
| FT4 | 10,7 pg/ml |
| Cortisol | 14 ug/dl |
| HCG | 0,05 |
| Densidad urinaria | 1,014 |
En el contexto de estudio del hipogonadismo se solicita cariotipo, el cual retorna 46, XX.
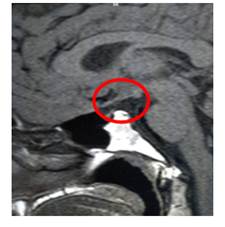
La resonancia magnética de hipófisis (Figura 1) informa anomalía hipofisaria congénita en relación a ectopia hipofisaria posterior, adenohipoplasia con tallo hipofisario fino en hilo, solo identificada tras la administración de contraste y un lóbulo posterior con brillo característico ubicado fuera de la localización habitual, inmediatamente superior al diafragma selar, hallazgos compatibles con Síndrome de Interrupción del Tallo Hipofisario.
Posterior al diagnóstico se reinicia sustitución de los ejes hormonales afectados.
DISCUSIÓN
El Síndrome de interrupción de tallo hipofisario es causa poco frecuente de hipopituitarismo2. Debido al número limitado de casos notificados, las manifestaciones clínicas de esta enfermedad son complejas y diversas. La ictericia neonatal prolongada, hipoglucemia, criptorquidia y micropene son comunes en las presentaciones de recién nacidos. Por otro lado, el retraso del crecimiento es la presentación más común en niños mayores y adultos4. La mayoría de los pacientes carecen de desarrollo sexual. Nuestra paciente había llegado a la edad adulta con baja estatura y falta de desarrollo de los caracteres sexuales. El Síndrome de interrupción de tallo hipofisario es una enfermedad predominantemente masculina, sin embargo, en este caso se trató de una mujer.
Se ha relacionado con formas severas de hipopituitarismo congénito, asociado a múltiples deficiencias de hormonas pituitarias, por lo que el diagnóstico generalmente se realiza en la infancia4, siendo infrecuente a edades tardías, como ocurrió en este caso.
El seguimiento estrecho de estos pacientes para el seguimiento de otras deficiencias hormonales es esencial si inicialmente se presentan con déficit de hormona de crecimiento aislado. En la literatura se reporta déficit de HGH en el 100% de los casos, FSH/LH en el 95,8%, ACTH en el 81,1% y TSH en el 76,3%7. En el caso presentado sólo se encontraron déficit de 2 ejes hipofisarios, el gonadotrofo y el somatotrofo.
Hallazgos como déficit en el crecimiento y pubertad retardada deben orientar a la realización oportuna de estudios de función hormonal y, de acuerdo con estos, valoración anatómica de la hipófisis en busca de la etiología.

