











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La autoinmunidad fue llamada con el término de “horror autotóxico” por Paul Ehrlich a principios del siglo XX(1). Con el paso de los años, este término mantiene el impacto acuñado por Ehrilch, y su mayor estudio ha permitido el descubrimiento de muchos otros mecanismos asociados al desarrollo de la misma. Actualmente se sabe que existen mecanismos defectuosos a nivel de la tolerancia central y periférica, involucrados en la etiología de las enfermedades autoinmunes, y se entiende que el proceso de autoinmunidad puede o no llevar al desarrollo de enfermedades autoinmunes, en dependencia de la exposición o no a estímulos ambientales y modificaciones epigenéticas(2).
La autoinmunidad se entiende como una respuesta inmunológica contra antígenos propios(3), cuyo desarrollo involucra defectos en el sistema inmune innato y adaptativo. Dentro de los mecanismos descritos, aquellos que se relacionan con el procesamiento y presentación de antígenos han llamado la atención, incluyendo defectos en el proceso de ubiquitinación y del funcionamiento del proteosoma. Entre las moléculas relevantes para el ensamble de proteosoma, POMP aparece como proteina fundamental para su adecuada función y ensamblaje, y sus alteraciones han mostrado estar relacionadas con una activación deficiente del proteosoma(4).
En este trabajo describimos las funciones del POMP y su relación con diferentes patologías autoinmunes e inflamatorias.
DEFINICIÓN
Las enfermedades autoinmunes son el resultado clínico de las diferentes alteraciones que ocurren en los mecanismos de tolerancia inmunológica. La autoinmunidad es el proceso por el cual la tolerancia inmunológica permite el desarrollo de células autoreactivas. Actualmente se han descrito diferentes mecanismos que conllevan a la autoinmunidad y sus enfermedades, como son el mimetismo molecular, la dispersión por epítope, y los epítopes crípticos. Estos mecanismos han sido descritos de manera individual, pero por sí mismos no son capaces del desarrollo y mantenimiento de los procesos patológicos autoinmunes. Por otra parte, la inmunidad innata ha mostrado defectos en diferentes niveles, los cuales pueden iniciar a nivel de la muerte celular y su aclaramiento, la presentación antigénica por células innatas, la activación de complemento, el funcionamiento de las células linfoides innatas, neutrofilos y la liberación de interferón. Por su parte, la inmunidad adaptativa ha sido estudiada en relación a la formación de anticuerpos y su consecutiva respuesta patológica, incluyendo aquellos mecanismos que alteran la tolerancia central y periférica(5-7).
Proteína POMP y el sistema de Ubiquitina-Proteosoma
La Proteína de maduración de Proteosoma (POMP) es codificada por el gene POMP localizado en el cromosoma 13 (13q12.3). También es conocida como “proteasemblina”, debido a su valiosa participación en el ensambleje del proteosoma(8).
El proteosoma participa en la importante función de la proteólisis, la cual en conjunto con el sistema proteosoma-ubiquitina y sistema autofágico lisosomal llevan a cabo toda la degradación proteínica humana. El proteosoma es un complejo enzimático de 2.5 Mda, formado de múltiples subunidades y su funcionamiento se ve reflejado también en la progresión del ciclo celular y la apoptosis. Está involucrado también en la transcripción, señalización y control de calidad proteínico, su trabajo lo realiza de forma conjunta con el complejo de ubiquitina, cuyo mecanismo consiste en el marcaje de proteínas y su direccionamiento al proteosoma. El complejo de ubiquitina, una vez formado, debe unirse a la subunidad 19S del proteosoma (complejo regulador), cuya regulación permitirá tomar el camino proteolítico y la liberación posterior de la ubiquitina. El complejo 19S está en contacto con el complejo 20S (complejo core), que a su vez, está formado por cuatro anillos, dos internos que corresponden a los anillos β y dos externos que corresponden a los anillos α. De este modo se forma el complejo 26S, cuyo nombre común es el de “proteosoma”, cuya ubicación se encuentra a nivel del citoplasma de las células eucariotas. Las proteínas que son consideradas sustrato para el proteosoma incluyen aquellas que participan en diferentes vías de señalización molecular, moléculas de supresión tumoral, factores de transcripción y moléculas inhibidoras, proteínas anti-apopotóticas y muchas otras más. El sístema de “anclaje” por ubiquitina, su direccionamiento al proteosoma y su adecuada lisis se conoce como el sistema “ubiquitina-proteosoma”, y sus defectos han sido relacionados con patologías autoinmunes e inflamatorias(9). Este sistema degrada principalmente proteínas solubles intracelulares, aunque también puede degradar proteínas transmembranales y endocitadas, incluso proteínas citosólicas digeridas por autofagia(10,11).
El sistema de ubiquitina está formado por 4 enzimas principales. Su proteína central es la ubiquitina, la cual se forma de 76 aminoácidos, las uniones de la ubiquitina están mediadas por el sistema enzimático de ligasas de ubiquitina y así se permite un fino proceso de anclaje y regulación de la proteólisis. El proceso de ubiquitinización pertenece a un proceso de modificación postraduccional permitido por la unión de un residuo de lisina de la proteína a eliminarse y la porción carboxil terminal de la ubiquitina. La ubiquitina cuenta con siete residuos de lisina, los cuales son todos útiles para su unión en la cadena de ubiquitina. El residuo K48 permite su direccionamiento al proteosoma. El proceso de ubiquitinización inicia con la enzima E1, la cual activa la ubiquitina por medio de un ATP, transfiriendo posteriormente esta proteina a la enzima E2, la cual a su vez es tomada por E3 y fijada a la proteina objetivo. Posteriormente, la ligasa E3 permitirá el incremento de la cadena de ubiquitina hasta ser catalizada por E4. La alta cantidad de genes conocidos para la ligasa de E3 sugiere la alta especificidad del proceso(11).
La proteina de maduración del proteosoma, POMP, es esencial para el ensamblaje del proteosoma, interactuando con la membrana del retículo endoplásmico y favoreciendo la unión de las unidades β, reclutando de este modo el anillo β y la posterior formación de la unidad 20S. Los sitios activos del proteosoma están representados por las subunidades β 1, β 2 y β 5, del complejo 20S, por lo que su inadecuado ensamblaje afectarían la adecuada acción de estos sitios activos. Estos sitios responderán con base en la estimulación de citocinas y la adaptación del proteosoma. Sin embargo, POMP no es la única proteina que permite un ensambleje adecuado, ya que algunas otras se encargan de la formación de este complejo “pre20S”. Dentro de las cuales se pueden considerar al complejo 13S y al complejo 16S. En el caso de POMP, se ha descrito su especificidad para la subunidad pro β 5. Dentro de las proteínas que participan en la síntesis del proteosona 20S, POMP no es la única, y en conjunto son conocidas como proteínas “chaperonas”, y en el caso de POMP, una vez cumplida su función de ensamblaje es eliminada. El complejo pre20S al igual que POMP y pro β5 están localizados en el retículo endoplásmico, lo cual ha sugerido a este como el principal sitio de formación del proteosoma(12).
POMP se considera como una proteína de membrana del retículo endoplásmico, cuya función no requiere algún otro complejo proteíco. Además, de su interacción con pre β5 también puede interactuar con algunas subunidades alfa, como alfa 3, 4 y 7. En particular, alfa 7 parece participar en el ensamblaje del anillo alfa del proteosoma. Su participación en la maduración del protesoma se considera como un modelo donde los anillos alfa son ensamblados por diferentes PACs (Proteinas chaperonas de ensamblaje), posteriormente el proceso final es llevado acabo principalmente en el retículo endoplásmico por medio de POMP(12,13).
ESTRUCTURA DEL PROTEOSOMA
El complejo 26S del proteosoma está formado por una unidad central 20S, con dos extremos correspondientes a una o dos unidades de regulación 19S. Estas unidades corresponden a los sitios de unión con el complejo de ubiquitina. En su interior, el complejo del proteosoma con forma cilíndrica continene 4 anillos, dos externos y dos internos. Los anillos externos, alfa, permiten junto con la unidad 19S la entrada de los sustratos al proteosoma, y los dos anillos internos, beta, conforman los canales catalíticos de los cuales se tienen 3 stios activos: el sitio de tipo quimiotripsina, el sitio de tipo tripsina y el sitio de tipo péptido hidrolasa post glutamil. Los anillos pentaméricos de tipo alfa pueden ser de 7 tipos (1 a 7), por su parte las subunidades beta pueden corresponder a dos de 7 tipos (1 a 7). Los anillos β actúan como hidrolasas nucleofílicas N-Terminal y utilizan sus residuos N-Terminal de treonina como los sitios activos. El complejo regulador 19S comprende a un anillo examérico de subunidades AAA-Atpasa y 12 subunidades no ATPasa. Esta porción reguladora, 19S, permitirá su unión con K48 de la ubiquitina, tomará la proteina “target”, y permitirá su ingreso a la unidad 20S. Otra porción reguladora no 19S es el complejo 11S, PA 200 y REG, los cuales tienen como objetivo brindar variabilidad al proteosoma. Esta variabilidad le dará al proteosoma gran heterogeneidad, la cual permanece en constante estudio. Algunos otros sitios funcionalmente importantes en la unidad 19S corresponden a los componentes de enzimas desubiquitinizantes, como la unidad intrínseca Rpn11 y las unidades extrínsecas Rpn1 y Rp13. En el caso de Rpn1 unira a K48. En el caso de Rpn11, su unión será dirigida a las cadenas de ubiquitina antes de su entrada a la porción ATPasa. Al salir del proteosoma, las proteinas serán degradadas a unidades aminoacídicas de 3 a 25 aminoácidos. Es relevante mencionar que algunas subunidades como B1, B3 y B5 son inducidas por interferón gamma sintético, y considerando que B5 es claramente inducido por POMP, la relación con interferón y POMP parece ser evidente(11,14).
Previo a la formación final de la estructura del proteosoma, POMP se encarga de prevenir la dimerización del proteosoma hasta que las unidades beta se encuentren posicionadas de manera adecuada, posterior a lo cual es degradado. A su vez, el proteosoma exige una regulación negativa altamente desarrollada, y en el caso de POMP, se ha descrito a miR-101, molécula supresora tumoral que se une al mRNA codificante para POMP, ocasionando una inhibición de ésta y un menor ensamblaje del proteosoma(15).
PAPEL DE POMP EN LA INFLAMACIÓN
Algunas patologías inflamatorias se han relacionado con alteraciones en POMP. Su asociación ha sido establecida con los defectos en el ensamblaje del proteosoma y su papel en la regulación y modulación de la respuesta inmune e inflamatoria. La actividad del sistema ubiquitina-proteosoma puede estar regulado por glucocorticoides esteroideos, citocinas y otros factores, incluyendo el interferón gamma (INFγ). Esta citocina puede regular las modificaciones de los sustratos para el proteosoma, como la familia de proteínas I Kappa B, y los componenentes enzimáticos de la maquinaria del sistema de ubiquitina, incluyendo la ligasa de ubiquiquitina Itch (Un tipo de ligasa E3 de ubiquitina). El inmunoproteosoma representa al proteosoma formado por una unidad reguladora diferente a 19S, siendo PA28 la característica del inmunoproteosoma. PA28 está formada por dos subunidades homólogas alfa y beta y relacionadas por PA28γ, conocida como antígeno Ki. Este inmunoproteosoma tiene una mejor capacidad para formar péptidos de unión al MHC clase I. A su vez el inmunoproteosoma es inducido por la expresión de INFγ. La mayoria de péptidos presentados por la vía de MHC I provienen del sistema del proteosoma. El sistema de ubiquitina- proteosoma también participa en la regulación del receptor de células T y la coestimulación con CD28, a través de la ligasa de ubiquitna CBl-β, cuya degradación elimina la regulación negativa para la expresión de citocinas proinflamatorias. Sin embargo, el principal mecanismo en la vía inflamatoria está en relación con NF-κB, cuya activación está mediada por el sistema ubiquitina-proteosoma. NF-κB es inhibido al unirse con IκB, el cual es eliminado por el proteosoma(16) (Figura 1).
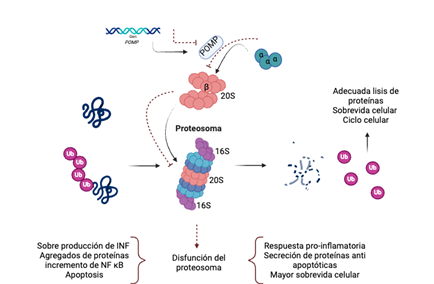
Nota. La función normal del proteosoma permite una adecuada proteólisis, asegurando un ciclo celular adecuado. Los defectos en el Gen POMP y su consecuente defecto en la proteína chaperona POMP pueden llevar a una disfunción del protesoma con el incremento en la producción de interferón, la acumulación de agregados proteicos, la activación de la vía de NF κB, defectos en la apoptosis, y aumento de la respuesta inflamatoria. Abreviaturas: POMP: Proteína de maduración del proteosoma. Ub: Ubiquitina. INF: Interferón. NF kB: Factor nuclear kappa B.
La relación entre POMP y el interferón es relevante en relación a la formación del inmunoproteosoma. Este complejo inmunológico se diferencia del proteosoma entre otras cosas por su expresión inducible, por lo que se ha llamado Proteosoma constitutivo (c20S) a aquel que permanece siempre expresándose sin importar el estímulo externo, a diferencia del inmunoproteosoma (i20S), cuya expresión ocurre bajo ciertos estímulos. Se ha reportado que el i20S se encuentra estimulado bajo el INFγ. Cuyo efecto está mediado directamente por POMP. En el trabajo realizado por Heink et al.(17), se relacionó el efecto sobre el i20S de POMP y la subunidad inmunológica Beta5i/LMP7 (Low molecular weigh protein 7) sobre la formación del inmunoproteosoma. INFγ induce directamente la biosíntesis de POMP y LMP7, con la consecutiva posición privilegiada de POMP para la biosíntesis del i20S. De igual manera, el silenciamiento de POMP ocasiona la fugaz aparición del proteosoma, lo cual permite generar un sistema de control eficiente y poco dañino. El INFγ también induce la formación de otras inmunosubunidades como LMP2 y MECL1. La mayor expresión de INFγ llevará entonces a la mayor expresión de POMP, LMP7 y una mayor generación de inmunoproteosoma. En condiciones específicas, este sistema favorece la respuesta inmune al cancer y la respuesta contra agentes infecciosos virales. Además de todo, la relación entre INFγ, POMP y LMP7 parece ser selectiva, ya que sus efectos parecen ser directos e independientes de otras citocinas presentes(17).
POMP EN LOS PROCESOS AUTOINFLAMATORIOS
La pérdida del control en el proceso de proteólisis, así como la alteración en los mecanismos involucrados es una característica de algunos procesos autoinflamatorios. Los defectos en el proteosoma se han asociado a síndromes inflamatorios nombrados como PRAAS (Proteasome associated autoinflammatory syndromes). Estos síndromes se acompañan por inducción de estrés oxidativo y activación del retículo endoplásmico. Es interesente que en estos síndromes se puede observar un incremento en los niveles de interferón. Como hemos mencionado antes, la estimulación por interferón puede llevar a la expresión del inmunoproteosoma, que a su vez estimulará nuevamente la vía de interferón hasta que el estímulo sea cebado. POMP, es también fundamental para el ensamblaje del inmunoproteosoma. Específicamente, la alteración del inmunoproteosoma se considera la causa de la agregación proteínica, y la liberación de interferón característica de PRAAS. Cada vez se han descrito más mutaciones asociadas al inmunoproteosoma, como aquellas que ocurren en las inmunosubunidades activas del proteosoma (B1, y B5), y en POMP, la cual ha sido descrita principalmente en el síndrome KLICK (Keratosis linearis with ichthyosis congenita and sclerosing syndrome)(18). La firma de interferón ha sido tan importante en algunos de estos síndromes, que han sido considerados como interferonopatías, como ocurre en el caso de PRAAS. La función de la firma del interferón en un contexto no viral aún se encuentra en estudio, sin embargo otra alternativa que se ha considerado es la estimulación no controlada de una infección viral inicial, cuyo final no puede suceder por la ausencia de un proteosoma funcional. Otros síndromes donde se ha descrito mutaciones en POMP, con firma de interferón son el Síndrome de JMP (Joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy), el Síndrome de Nakajo-Nishimura y el Síndrome de CANDLE (Cronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature)(19).
El síndrome KLICK se considera un síndrome autoinflamatorio secundario a una mutación en POMP, específicamente se ha propuesto una deleción del nucleótido 1bp en los queratinocitos diferenciados, lo que ocasiona una insuficiencia del proteosoma. Estos niveles bajos de POMP ocasionan el estado inflamatorio predominante en la piel. El síndrome KLICK se caracteriza por la aparicion congénica de una ictiosis generalizada, queratoderma con esclerosis palmoplantar, deformidades, y múltiples pápulas. En 1997 se propuso el acrónimo KLICK, considerándose de manera inicial un síndrome de herencia autosómica recesiva(20). Usando un screen genético, se demostró una deleción de un nucleótico único en la porcion UTR 5´del gen POMP en 12 pacientes con síndrome de KLICK, así como su relación con los hallazgos cutáneos y su consecutiva afectación al proteosoma(21). Hasta hace algunos años, pocos casos con esta mutación habían sido descritos. Los datos clinicos cutáneos en KLICK se consideran secundarios a los procesos inflamatorios que resultan de la estimulación por stress del retículo endoplásmico, el cual resulta de la insuficiencia del proteosoma. Esta insuficiencia se asocia con la estimulación del interferón y la acumulación proteínica(22-24).
Además del síndrome de KLICK y PRAAS, diversas otras enfermedades se han asociado a mutaciones en POMP. Algunos síndromes autoinflamatorios asociados al proteosoma y POMP se han descrito como PRAAS2 o PRAID (POMP-Related autoinflammation and immune dysregulation disease). Con respecto a PRAID, sin embargo, se considera una fisiopatología distinta, cuya disregulación inmunológica está asociada a un efecto negativo secundario a los defectos en POMP. La primera mutación reporatada en PRAAS 2 se reportó en el exon 5, ((mutación c.344_345insTTTGA (p.Glu115Aspfs*20)), la cual ocasiona haploinsuficiencia y deficiencia en la actividad del proteosoma. Posteriormente, se reportaron dos pacientes no relacionados con mutaciones distintas ((c.334_335delAT (p-I1e112Trpfs*3)) y ((c.342_348delinsACC(p.Phe114Leufs*18)), en cuyos casos se propuso el mecanismo de estimulación negativa, debido a un proceso de escape y decaimiento no mediado por RNAm, resultando en un ensamblaje perturbado del proteosoma. Y consideran que este proceso puede llevar a una disregulación inmunológica que involucra la autoinflamación y la autoinmunidad. Ambos casos reportados presentaron un cuadro caracterizado por inmunodeficiencia, inflamación y autoinmunidad. En ambos casos reportados, se demostró también una asociación con la liberación incrementada de interferón y la mayor expresion de estrés en retículo endoplásmico(25). Estos casos, sin embargo, no son los primeros que se asocian a una inmunodeficiencia. Gatz et al. (26), describieron un paciente pediátrico con mutaciones en POMP y MCM3AP con una inmunodeficiencia y síntomas cutáneos, considerando que el efecto de la mutación en POMP estaba relacionada a su impacto en la maduración del proteosoma, y la vía de señalización de NFκB, que al estar disminuida comprometía la función de linfocitos T y B.
Algunos otros síndromes autoinflamatorios con predilección por la piel, específicamente, caracterizados por un proceso patológico de queratinización han sido descritos con el término de AiKDs (Autoinflammatory Keratinization diseases). Este término incluye procesos autoinflamatorios ocasionados por diferentes vías, que pueden incluir, las alteraciones en el inflamasoma, la vía de NF kB, el interferón y otro grupo de alteraciones misceláneas, incluyendo a POMP. Estas enfermedades incluyen a enfermedades como la psoriasis pustular generalizada, pustulosis palmoplantar, impétigo herpetiforme, pitiriasis rubra pilaris, acromermatitis contínua, CARD14-asociada a erupción papuloescamosa y otras más(27).
POMP EN AUTOINMUNIDAD
Como ya sabemos, el principal rol del Gen POMP corresponde a la maduración del proteosoma, este último encargado principalmente de reciclar y degradar proteínas innecesarias, jugando así un rol importante en la homeostasis. Recordando un poco la fisiopatología de algunas enfermedades autoinmunes tales como el lupus eritematoso sistémico (LES), ésta se centra en el mal aclaramiento de residuos apoptoicos, creando asi auto-anticuerpos que reaccionan a dichas proteínas(28), de tal forma que, asumiendo y conociendo el rol que tiene POMP en la homeostasis celular, una mutación a este nivel podría condicionar la aparición del mal aclaramiento apoptóico, aunado a que si el paciente presenta factores predisponentes o bien, está expuesto a ciertos factores ambientales que pudieran jugar un rol detonador, podríamos deducir que el desenlace sería el desarrollo de alguna enfermedad autoinmune. Por otra parte, en el LES y otras enfermedades autoinmunes está demostrada la relación entre los defectos de la inmunidad innata y algunas citocinas relacionadas, sobresalinedo INF(29,30).
Como se ha comentado antes, el papel del interferón ha sido estudiado en relación con alteraciones en la expresión de Gen POMP, cuya expresión puede ser incrementada o disminuida, con base en diferentes mecanismos inmunológicos, como lo describió Poli et al, en el síndrome de disregulación inmune con inmunodeficiencia y autoinmunidad(25). Además de los defectos en el aclaramiento apoptótico, el incremento en los niveles de interferón, los mecanismos defectuosos en la inmunidad innata y los autoanticuerpos, también se han relacionado los defectos de autoinmunidad, como el sistema de ubiquitinización(31). Para estos diferentes mecanismos descritos es relevante el papel de la proteína POMP.
Por parte de nuestro equipo, se describió un caso de un paciente pediátrico con datos clínicos sugestivos de LES, cuyo comportamiento clínico agresivo no permitió su estudio a profundidad. Sin embargo, presentó una buena respuesta inicial a la terapia inmunosupresora, lo cual sugería un proceso inflamatorio sistémico autoinmune. Este paciente presentó una deleción en el Gen codificante para la proteína POMP(32).
También se han descrito alteraciones del sistema de proteosoma y ubiquitina en otras enfermedades autoinmunes, como Esclerosis Múltiple, Artritis Reumatoide y algunas miopatías, asociándose con presencia de autoanticuerpos contra algunas subunidades del proteosoma(33-34).
CONCLUSIÓN
Los mecanismos inmunológicos en los que la proteína POMP se ve involucrada son variados. Su papel en la maduración del proteosoma e inmunoproteosoma es el principal. Sin embargo, cada vez existe más evidencia sobre las alteraciones que puede ocasionar derivando en diversas enfermedades que van desde enfermedades inflamatorias, autoinflamatorias, autoinmunes e inmunodeficiencias. Consideramos que los defectos en POMP representan otro camino hacia la autoinmunidad, y su mayor estudio permitirá este entendimiento.

