











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La diversidad patogénica del Staphylococcus aureus resistente a meticilina (SARM), manifestada por su destreza para colonizar, adaptarse y sobrevivir en distintos tejidos celulares durante la infección, se debe principalmente a la portación de genes que le confieren resistencia a los antibióticos, como la resistencia a meticilina que está determinada por el gen cromosómico “mecA”, así como genes codificantes de factores de virulencia que contribuyen con su patogenicidad1.
La determinación de la variabilidad genética de los aislamientos SARM, necesaria para la comprensión de la expansión y diseminación bacteriana, así como para establecer las estrategias necesarias para la limitación de su propagación, sólo pueden realizarse mediante el empleo de métodos moleculares combinados, debido a su gran complejidad genética. Entre los métodos que permiten establecer relaciones evolutivas entre clones se encuentran, aquellos basados en el estudio de restricción del genoma completo como la Electroforesis en Gel de Campo Pulsado (PFGE), considerada como “gold estándar”; las técnicas basadas en amplificación de múltiples VNTR (Regiones de Número Variable de Repeticiones en Tándem) como la MLVA, así como las basadas en la secuenciación de locus hipervariables, que incluyen la tipificación de secuencias multilocus (MLST, por la sigla en inglés de (Mutilocus sequence typing) y la tipificación del gen de la proteína A (Spa typing), entre otros2-5.
En Paraguay existen algunos estudios sobre la variabilidad genética de SARM6,7, sin embargo, queda mucho por estudiar respecto a la epidemiología de esta bacteria, para lo que la implementación de metodologías de identificación clonal de rápida respuesta y accesibles a nivel nacional resulta esencial. En el 2017 nuestro grupo de investigación incursionó de forma exitosa con la utilización de la MLVA manual para la tipificación de S. aureus obteniendo excelentes resultados comparativos a la PGFE8, lo que ha permitido analizar aislamientos SARM y generar los primeros perfiles de agrupación en base a similitud genética de ADN aplicada al análisis de fragmentos, siendo una herramienta alternativa que permitió mejorar el tiempo de respuesta en la resolución de posibles casos de brotes. Sin embargo, debido al procesamiento manual de las muestras, esta técnica presenta como limitación la generación de resultados en un tiempo prolongado para evaluar múltiples aislamientos.
En este contexto el presente estudio tuvo como principal objetivo el análisis de variabilidad genética de SARM-AC (Staphylococcus aureus resistentes a meticilina aislado de la comunidad) y SARM-AH (Staphylococcus aureus resistentes a meticilina aislado del hospital) que produjeron infecciones invasivas en niños paraguayos durante los años 2010, 2012 y 2013 mediante la estandarización y aplicación del método MLVA en su formato automatizado, siendo una tecnología de punta para estudios de brotes, y por tanto un aporte hacia la mejor comprensión de la epidemiología de las infecciones invasivas causadas por SARM en nuestro país.
MATERIALES Y MÉTODOS
Diseño del estudio y Población
Se analizaron 25 cepas SARM, representantes de más de 700 aislamientos de S. aureus colectados en los años 2010, 2012 y 2013 de 4 hospitales de referencia del departamento Central de Paraguay, participantes del estudio (Hospital General Pediátrico Niños de Acosta Ñú, Hospital Central del Instituto de Previsión Social, Hospital de Clínicas y el Hospital Nacional de Itauguá), todos provenían de muestras de niños con edades comprendidas entre 1 y 16 años. Los aislamientos analizados se encontraban criopreservados en BHI-glicerol en el Biobanco del IICS, con ficha de identificación y antibiograma correspondiente, así como datos clínicos mínimos del paciente a quien pertenecían. La colecta de muestras se realizó durante los años mencionados como parte de proyectos marcos existentes en el grupo de investigación de S. aureus.
Los aislamientos incluidos para el análisis fueron aquellos considerados resistentes a meticilina según los criterios del CLSI (Clinical and Laboratory Standars Institute)9-11, obtenidos a partir de cultivos de muestras biológicas de niños como sangre, secreción purulenta, líquido articular y secreción traqueal, consideradas como provenientes de infecciones invasivas, en alguno de los cuatro centros hospitalarios de referencia participantes del estudio. Además contaban con genotipificación previa que incluía la detección del gen de resistencia a meticilina (mecA) y codificantes de factores de virulencia (pvl, sea, seb, sec, sed, seh, hla, hlb, etA y etB)12-15, como también del análisis de tipificación molecular por MLVA manual8, Spa typing5 y el análisis de PFGE. Los aislamientos analizados eran representativos de perfiles PFGE obtenidos en estudios previos16 .
Los aislamientos criopreservados se repicaron en medio TSA (Tripteína Soya Agar) e incubadas a 35°C por 24-48hs en atmósfera de 5% de CO2, para la posterior extracción del ADN genómico empleando dos métodos distintos con la finalidad de comparar la eficacia: kit comercial de purificación de ADN genómico (Wizard Genomic, Promega, EEUU) y kit de extracción de ADN genómico en columna (QIAamp DNA Mini Kit, Qiagen, EEUU) siguiendo las instrucciones de los fabricantes. Posterior a la extracción el ADN fue conservado a -20°C y para la cuantificación de este se realizaron lecturas de absorbancia a 260nm utilizando el cuantificador de ADN NanoDrop (ND-2000).
Estandarización de la MLVA
Se amplificaron 6 regiones VNTR Sa022, Sa0704, Sa1132, Sa1291, Sa2039 y Sa2511, según lo descrito por Sobral et al.17, empleando oligonucleótidos marcados con fluorescencia, sintetizados por las empresas Applied Biosystem (EEUU) y Macrogen (Corea), donde para cada sistema el oligonucleótido “forward” fue marcado con un fluoróforo en el extremo 5´.
Para la estandarización de las reacciones de PCR se realizaron varias pruebas con variaciones de calidad y cantidad de ADN (20ng/µL, 40ng/µL, 80ng/µL), concentración de cloruro de magnesio (2,5mM, 2,8Mm, 3mM) y condiciones de ciclado en la que se probaron diferentes temperaturas de anillamiento. En base a la intensidad de los productos durante la amplificación se seleccionaron los sistemas que resultaron las mejores condiciones de reacción y ciclado de la PCR para ser detectados por fluorescencia.
Se verificó la amplificación por PCR de los fragmentos analizados mediante electroforesis en gel de agarosa al 2% y tinción con Syber safe al 10%, previo al envío de productos para análisis automatizado (Macrogen, Corea).
Análisis de MLVA por electroforesis capilar
Los productos de PCR de los 6 sistemas fueron remitidos a Macrogen (Corea) para el análisis de electroforesis capilar con el analizador de ADN Applied Biosystems® 3500 Genetic Analyzer.
Obtención de dendrograma
Los datos generados por la electroforesis capilar se emplearon para la generación de un dendrograma mediante el software FigTree V.1.4.0. Las agrupaciones fueron numeradas para su identificación, según recomendación de Sabat et al. en el 20033, en base a su similitud. Se efectuó la comparación de patrones MLVA obtenidos para establecer existencia de proximidad evolutiva.
Análisis Estadístico
Los datos de los aislamientos obtenidos de las fichas epidemiológica, microbiológica y molecular fueron introducidos y almacenados en una planilla electrónica y los cálculos estadísticos fueron realizados empleando los softwares Microsoft Excel y Epiinfo Versión 7.2.2.6. Se empleó estadística descriptiva para referir las características de la población y los perfiles de MLVA automatizados analizados.
RESULTADOS
De los 25 aislamientos que causaron infecciones invasivas en niños, el 64% corresponde a SARM adquiridos en la comunidad (SARM-AC) y el 16% restante corresponde a SARM adquiridos en el hospital (SARM-AH). La tendencia de distribución por franja etaria fue similar en los cuatro hospitales participantes del estudio. Los aislamientos fueron obtenidos de muestras de sangre (60%), secreciones purulentas (32%), líquido articular (4%) y secreción traqueal (4%), y las infecciones invasivas causadas por estos aislamientos incluyeron neumonía bilateral, sepsis neonatal, artritis séptica, osteomielitis, sepsis con foco abdominal o articular, absceso parietal, entre otras.
En cuanto al efecto de la calidad de ADN se observaron mejores resultados evidenciados por una mayor cantidad de producto obtenido utilizando 20ng/µL de ADN molde extraído por el kit de extracción en columna (Qiagen, USA).
Respecto al efecto de la concentración de cloruro de magnesio se observó que las reacciones de PCR para los sistemas Sa2511, Sa0704 y Sa1291 presentaron mejores resultados utilizando 2,8mM de MgCl2 por tubo de reacción. Sin embargo, para los sistemas Sa0266, Sa2039 y Sa1132 la concentración óptima resultó 3mM por tubo de reacción.
Las condiciones de amplificación testadas se realizaron con variaciones en la temperatura de anillamiento respecto a los 6 sistemas VNTR. Para los sistemas Sa0704, Sa1291, Sa2039 y Sa1132 la temperatura de anillamiento que presentó mejores resultados fue de 51°C, mientras que la temperatura de anillamiento óptima para los sistemas Sa0266 y Sa2511 fue de 50°C y 54°C respectivamente.
El ADN se amplificó por sistemas individuales que comprendían 6 loci descritos previamente. Los productos de cada aislado fueron remitidos a Macrogen agrupados en dos tubos en base a la combinación de fluoróforos y los rangos de tamaño de productos de amplificación esperados, el primero conteniendo los sistemas Sa1132, Sa0704, Sa2039, Sa2511 y Sa1291, y el segundo tubo para el sistema Sa0266, para la electroforesis capilar.
Del análisis de los electroferogramas correspondientes para cada aislamiento se observó 100% de eficiencia en la detección de productos compatibles con tamaño y marcación fluorescente en los sistemas Sa1132, Sa0704 y Sa2511, 96% en Sa2039 y Sa1291 y 88% en Sa0266.
Agrupaciones resultantes de análisis de MLVA manual
En el análisis de la MLVA manual previamente estandarizada y validada con PFGE y spa typing por nuestro Grupo de investigación de S. aureus, se logró la amplificación total de 7 loci VNTR (cflA, cflB, sdrC, sdrD, sdrE, Spa, SspA) de los 25 aislamientos SARM en estudio, agrupados en 4 perfiles diferentes, Perfil 1-t019 (76%), Perfil 2-t311 (12%), Perfil 3-t002 (8%) y Perfil 4-t11770 (4%)8) .
Agrupaciones resultantes del análisis por MLVA automatizado
Del análisis por MLVA automatizado, 20 aislamientos (20/25, 80%) generaron productos en todos los loci (n=6). De los 5 aislamientos que no generaron productos en algunos de los loci fueron excluídos 3 aislamientos del dendrograma, por no poder ser analizados según criterios internacionales de agrupación de aislamientos (Malachowa et al., 2005). Pudiendo incluirse en el análisis informático a 22 aislamientos18.
De los productos amplificados de los 6 sistemas VNTR de los 25 aislamientos SARM que fueron enviados a Macrogen (Corea), los sistemas Sa1132, Sa704 y Sa2511 resultaron los más eficientes, detectándose fragmentos en el 100% de los aislamientos, seguidos por los sistemas Sa2039 y Sa1291 en los que fueron detectados productos con una eficiencia del 96% y por último el sistema Sa0266 con 88% de eficiencia, presentando en uno de los aislamientos una banda secundaria, por lo que resultó ser el sistema menos eficiente. La distinción de la banda secundaria pudo discriminarse mediante la intensidad de los picos que se generaron en el análisis por electroforesis capilar, siendo mayor el valor de intensidad del pico de la banda primaria en comparación con la secundaria.
El análisis del patrón de bandas, así como la comparación de bandas comunes por la técnica MLVA automatizada permitió distinguir 3 perfiles o agrupaciones diferentes, siendo el Perfil 1-t019 más frecuente (86%), seguido del Perfil 3-t002 (9%) y Perfil 2-t311 (5%).
Los dendrogramas tanto el de referencia, obtenido por el método manual, y el obtenido por el método automatizado se muestran en la Figura 1. La comparación de las agrupaciones por ambos métodos arrojó un nivel de concordancia del 100% con un índice Kappa igual a 1,0 en los 22 aislamientos SARM que fueron incluidos para la comparación.
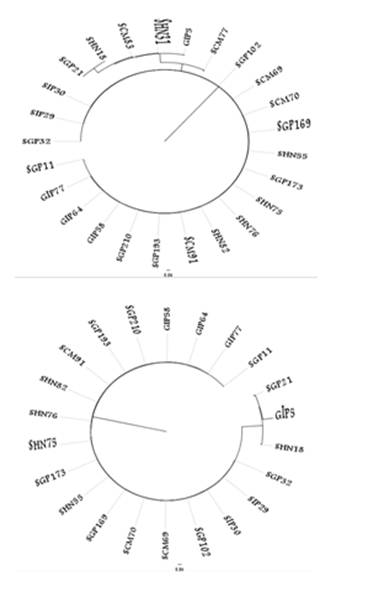
Figura 1: Agrupaciones de SARM causantes de infecciones invasivas. Dendrogramas obtenidos con “software” FigTree. A) Dendrograma a partir de método manual: Se visualizan 4 perfiles diferentes por el método de MLVA manual en 25 aislamientos SARM, siendo el Perfil 1-t019 más frecuente (n:19), seguido del Perfil 2-t311 (n:3), Perfil 3-t002 (n:2) y Perfil 4-t11770 (n:1), B) Dendrograma a partir de método automatizado: Se visualizan 3 perfiles diferentes por el método de MLVA automatizado en 22 aislamientos SARM, siendo el Perfil 1-t019 más frecuente (n:19), seguido del Perfil 3-t002 (n:2) y Perfil 2-t311 (n:1).
DISCUSIÓN
La circulación regional y mundial de clones SARM exitosos, incluso epidémicos ha impulsado el desarrollo de herramientas moleculares precisas para el abordaje de la compleja epidemiología de este patógeno. En este aspecto uno de los brotes más graves registrados en Paraguay por S. aureus fue la intoxicación por consumo de leche ultrapasteurizada contaminada con S. aureus portador de enterotoxinas C y D en el cual resultaron intoxicadas 400 personas, de las cuales 60 requirieron hospitalización, registrándose incluso el óbito de una lactante menor, donde la investigación del brote desde el aspecto molecular tuvo que realizarse en el exterior en el 201119; poniendo de manifiesto la imperiosa necesidad de implementar métodos moleculares sensibles pero de baja complejidad y fácilmente accesibles en Paraguay. En este contexto este grupo de investigación de S. aureus inició la puesta a punto, validación y empleo de la técnica MLVA, en formato manual, que si bien arrojó excelentes resultados en comparación con el estándar de oro (PFGE), presentaba el inconveniente de requerir un tiempo de análisis extendido y permitir el análisis de un número limitado de aislado debido al empleo de técnicas moleculares un tanto engorrosas8.
En este punto la inclusión de metodología automatizada de análisis de fragmentos se planteó para ampliar la capacidad de análisis de variabilidad genética de SARM y la disminución del tiempo de respuesta en la generación de resultados. La automatización de la técnica MLVA siguiendo el protocolo descrito por Sobral et al.17, requirió numerosas modificaciones ante la imperiosa necesidad de lograr el mayor rendimiento en cantidad de producto de PCR y disminuir en lo posible los artefactos como bandas secundarias y dímeros de oligonucleótidos. El conjunto de ajustes realizados permitió la tipificación por MLVA automatizado del 88% de los aislados analizados, obteniendo un rendimiento equiparable a otros estudios internacionales que reportan niveles de tipificación entre el 76 a 100%. Así mismo, el evento de no lograr la amplificación de la totalidad de los locus, que origina perfiles parciales es un hecho descrito por diversos autores y cuya causa podría asociarse a la presencia de mutaciones en las regiones de anillamiento de los oligonucleótidos3,18,20,21.
La comparación entre los dos métodos mostró excelente concordancia, indicando una fuerza de correlación perfecta entre estos dos métodos de tipificación genética, en donde se obtuvieron 3 perfiles MLVA distintos para los 22 aislamientos SARM que fueron comparados. Esta elevada correlación entre estos métodos ya fue demostrada en varios estudios como el realizado por Sobral et al en el año 201217.
El método MLVA automatizado presenta alta concordancia con el método MLVA manual que ya fue validada con el PFGE, por lo que la concordancia de la MLVA automatizada con el método “gold estándar” (PFGE) se mantiene, como también con otras técnicas ampliamente utilizadas como la tipificación de locus de la proteína A y MLST.
Este tipo de metodología de complejidad instrumental no sólo permite llevar a cabo la caracterización de aislamientos con alta sensibilidad y un óptimo tiempo de respuesta en comparación con el método manual sino que también podría optimizar el uso de recursos en el empleo de PFGE que hasta ahora es muy restringido en Paraguay. A futuro podrían ajustarse condiciones para una amplificación múltiple y cuya marcación con fluoróforo permita el análisis por electroforesis capilar en un solo tubo por muestra, reduciendo tanto costos como tiempo de generación de resultados.
La técnica de análisis automatizado de fragmentos tiene muchas aplicaciones como en la investigación genómica, diagnóstico molecular, clínica y farmacéutica, sin contar con las aplicaciones forenses22. La rapidez del análisis, conjugada con la automatización del sistema, colocan al análisis automatizado de fragmentos como una herramienta indispensable en el área diagnóstica23, por tanto la experiencia que describimos puede ser de utilidad para otros grupos nacionales interesados en el uso de esta técnica.
En este trabajo se analizaron 25 aislamientos que son representativos de más de 700 aislamientos de S. aureus provenientes de 4 hospitales distintos, ha permitido señalar como mayoritario al Perfil 1-t019, relativo al clon SARM ST30-t019-IV-PVL, como el clon predominante causante de infecciones invasivas en niños asociadas a SARM en el país, concluyendo que la elevada prevalencia del mismo podría deberse a una ventaja adaptativa que permita desplazar a los demás clones, ya que se está instalando en el país en varios hospitales y en la comunidad, facilitando aún más su diseminación24) . En efecto, el mismo es considerado como un clon pandémico en Sudamérica, ya que existen estudios previos que lo reportan en Chile, Argentina, Colombia y Paraguay25. En segundo lugar el Perfil 2-t311 (12%), relacionado al clon SARM ST5-t311-IV-PVL y portador del gen codificante de la enterotoxina A (sea), descrito como causante de infecciones severas en la comunidad13,19,26-28. Además, otro estudio argentino realizado por Egea y colaboradores, resultaron como “spatipos” más frecuentes relacionados a infecciones por SARM, el t019 (45%) y el t311 (45%). Estos resultados, agregados a los generados por este estudio, muestran un gran predominio y amplia distribución de estos dos clones a nivel regional29,30.
La baja frecuencia del Perfil 3-t002 es similar a lo reportado en Argentina en el año 2018 en un estudio de aislamientos S. aureus en adultos hospitalizados31, lo que hace que el mismo sea considerado como esporádico. Además un aislamiento fue categorizado como el Perfil 4-t11770, relacionado con el clon SARM ST8-IV-t11770-PVL que es genéticamente similar a la Variante Latinoamericana USA300 que es un clon hipervirulento y epidémico frecuente en la región norte y sur de Sudamérica32.
CONCLUSIONES
El clon SARM predominante hallado en este estudio fue el de Perfil 1-t019, seguidamente de los clones de Perfil 2-t311, Perfil 3-t002 y Perfil 4-t11770. La agrupación por el método MLVA automatizado presentó excelente concordancia con el método MLVA manual y el PFGE, además nos permitió ampliar la capacidad de análisis de variabilidad genética de SARM, mejorando la respuesta en la generación de resultados.
La técnica tiene un gran potencial en el estudio de brotes hospitalarios e intoxicaciones alimentarias causadas por S. aureus, así como es una herramienta muy interesante útil para la aplicación en otras áreas de salud en el país.

