











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El virus del papiloma humano (VPH), es un virus no envuelto, su genoma está compuesto por ácido desoxirribonucleico (ADN) circular de doble hebra, cuyo tamaño es 8000 nucleótidos aproximadamente1.
El genoma del VPH está formado por 3 regiones: LCR (Long control region), región no codificadora que posee secuencias que controlan la transcripción y replicación viral. Región temprana: contiene 6 marcos de lectura abiertos (Open Reading Frame-ORFs) que contienen las secuencias codificantes de las proteínas E1, E2, E4, E5, E6 y E7, involucradas en múltiples funciones incluyendo la activación de la transcripción, transformación, replicación y la adaptación viral a diferentes ambientes celulares. Región tardía: contempla a los ORFs que contienen la secuencia codificante de las proteínasL1 y L2 de la cápside viral, las mismas forman la estructura del virión y facilitan el empaquetamiento del ADN1.
El VPH pertenece a la familia Papillomaviridae y se clasifica según la identidad de la secuencia de nucleótidos del gen L1, en género, especie, tipo, subtipo y variantes. La identificación de los tipos virales de VPH se basa en el marco de lectura abierto de L1 debido a que es la región más conservada en el genoma viral. Un tipo es considerado nuevo si el genoma completo es clonado y la secuencia completa de nucleótidos del gen L1 difiere en más de 10% del tipo viral conocido más cercano. Una diferencia entre 2 y 10% define un subtipo, y una diferencia menor a 2% define a una variante2.
El VPH está asociado a diferentes patologías, desde lesiones benignas a tumores invasivos. Los VPH mucosos del género alfa-papillomavirus, son VPH oncogénicos, responsables de la mayoría de los casos de cáncer de cuello uterino. Por otro lado, también son los responsables de otros tipos de cánceres como el anal, vulvar, de pene, vaginal y orales, pero en menor proporción3.
El VPH 16 pertenece al género alfa-papillomavirus4. El mismo es el tipo viral más frecuente en mujeres con citología normal, lesión de alto grado y con cáncer de cuello uterino invasivo a nivel mundial5-7. Es el responsable de aproximadamente el 60% de los casos de cáncer de cuello uterino a nivel mundial7. En nuestro país el VPH 16 es el más frecuente tanto en mujeres con cáncer de cuello uterino, así como también en mujeres con y sin lesión de cuello uterino8-11.
Las variantes de VPH 16, a la fecha se clasifican en 4 linajes y 16 sublinajes: linaje A, incluye los sublinajes A1, A2, A3 (anteriormente conocidas como europeas) y A4 (asiática), linaje B, sublinajes B1, B2, B3 y B4 (africana-1), linaje C, sublinajes C1, C2, C3 y C4 (africana-2), linaje D, sublinajes D1 (norteamericana, NA), D2 y D3 (asiático-americana, AA) y D412.
Una variante de un tipo viral conocido se considera cuando el aislado viral difiere en menos del 2% de la secuencia de nucleótidos del gen L1. La secuenciación de la región LCR se utiliza con frecuencia para clasificar la diversidad intra-típica y los linajes de las variantes, debido a que esta región es la menos conservada y es la que presenta mayor variabilidad entre los diferentes genomas de VPH. Además, la misma controla la transcripción y replicación viral mediante la unión de factores de transcripción a la misma. Las diferentes variantes de VPH 16 presentan diferencias en esta región, implicando diferencias en la unión de factores de transcripción, determinando así la funcionalidad1,13.
La secuenciación completa del gen E6 junto con la región LCR es utilizada a fin de que las mismas sean comparables con la clasificación de las variantes en base a la secuenciación completa del genoma del VPH 1614.
Con respecto a las variantes de VPH 16 y la patogénesis cervical, existe evidencia suficiente que demuestran que la persistencia, progresión a lesiones precancerosas, desarrollo de cáncer, así como también el tipo de cáncer cervical se encuentran relacionados a la misma15.
Por ello, el objetivo del presente trabajo fue optimizar dos reacciones de amplificación en cadena de la polimerasa (PCR) que abarcan a la región no codificante LCR y al gen E6 cuyos productos podrán ser sometidos a secuenciación por el método de Sanger. El análisis de la secuencia nucleotídica de estas regiones del genoma servirá para determinar las variantes de VPH 16. Por otra parte, las secuencias obtenidas se utilizarán para construir plásmidos, pudiendo posteriormente evaluar la actividad transcripcional de la región no codificante LCR de las diferentes variantes de VPH16 a través de la medición indirecta de la actividad de la luciferasa de vagalume en ensayos de transfección, con el fin de relacionarlas con la patogénesis cervical.
MATERIALES Y MÉTODOS
Para la optimización de las PCR se utilizaron muestras de VPH 16, previamente tipificadas (Proyecto código 14-inv-036), provenientes del biobanco del Departamento de Salud Pública.
Extracción de ADN viral
Para la extracción del ADN viral se utilizó el kit comercial GeneJet Genomic DNA Purification Kit (Thermo Scientific, EEUU) y se siguió el protocolo proveído por el fabricante. El ADN extraído fue almacenado a -80°C hasta su procesamiento.
Optimización de las PCR
Para la optimización en la amplificación de la región LCR y del gen E6 se utilizaron los cebadores descriptos por Xi et al 2017 y Cornet et al 2012, respectivamente 13,14 (Tabla 1).
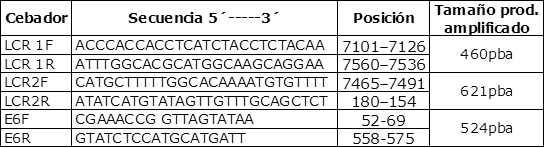
Se probaron tres PCR. Cada una utiliza un par de cebadores que amplifican la región 5´de LCR (cebadores LCR1), la región 3´ de LCR hasta la región 5´de E6 (cebadores LCR2) y por último cebadores para las regiones 5´y 3´de E6 (cebadores E6) (Tabla 1). Cada amplicón posee un extremo que se superpone con el subsiguiente, por lo que con el empalme de los tres productos se obtiene una secuencia consenso desde la posición 7101 (LCR) a 575 (E6) del genoma circular de VPH 16. Todas las PCRs fueron probadas siguiendo las recomendaciones del fabricante de la enzima ADN polimerasa utilizada de alta fidelidad, (iProof High-Fidelity DNA Polimerase,Bio-Rad), considerando que los productos de amplificación serán secuenciados.
Cada reacción se realizó en un volumen final de 50uL con concentraciones finales 1X de buffer, 1,5mM de MgCl2, 200uM de dNTP, 0,2uM de cada cebador y 0,02U/uL de iProof DNA polimerasa.
Teniendo en cuenta la temperatura de melting (Tm) de cada par de cebadores, cada PCR se probó a diferentes temperaturas de anillamiento. Además, se probaron los diferentes buffers proveídos con la enzima. Para la PCR con los cebadores LCR1 y LCR2 se probaron las temperaturas de 54 ºC, 57 ºC y 60ºC. Para los cebadores E6 se probaron las temperaturas de 48ºC, 50 ºC y 52ºC. En la tabla 2 se describe el programa de termociclado utilizado según las recomendaciones del fabricante de enzima ADN polimerasa utilizada.
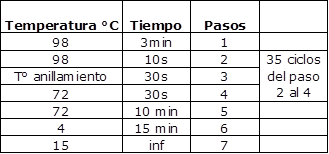
Los productos de amplificación fueron sometidos a corridas electroforéticas en gel de agarosa al 1%. Los productos amplificados fueron visualizados mediante tinción y revelado. Para la tinción se utilizó el colorante Diamond ™(Promega, EEUU).
RESULTADOS
Optimización de la PCR LCR1 y LCR2
Con respecto a la temperatura de anillamiento, se observó producto de amplificación en las 3 temperaturas probadas, sin embargo, se observó una mayor intensidad al realizar la PCR a 57°C utilizando el buffer GC, que incrementa la efectividad para secuencias con alto contenido de bases G-C. En las Figuras 1-3 se observan las corridas electroforéticas de productos amplificados obtenidos en las diferentes PCRs utilizando diferentes temperaturas de anillamiento y buffers de reacción.
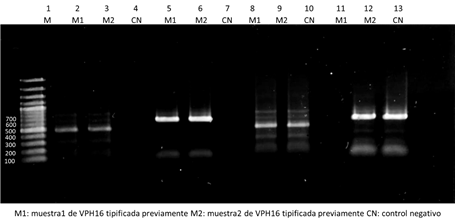
Figura 1. Corrida electroforética en gel de agarosa al 1% de productos amplificados de VPH 16 con cebadores LCR1 y LCR2 utilizando una temperatura de anillamiento de 54ºC. Carril 1: Marcador de pares de base. Carriles 2-7 se utilizó buffer HF y del 8-13 buffer GC. En los carriles 2, 3, 8 y 9 se sembraron productos amplificados de 460 pb, obtenidos mediante cebadores LCR1. En los carriles 5, 6, 11 y 12 se sembraron productos amplificados de 621 pb obtenidos mediante cebadores LCR2.
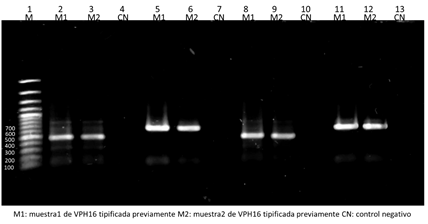
Figura 2. Corrida electroforética en gel de agarosa al 1% de productos amplificados de VPH 16 con cebadores LCR1 y LCR2 utilizando una temperatura de anillamiento de 57ºC. Carril 1: Marcador de pares de base. Carriles 2-7 se utilizó buffer HF y del 8-13 buffer GC. En los carriles 2, 3, 8 y 9 se sembraron productos amplificados de 460 pb, obtenidos mediante cebadores LCR1. En los carriles 5, 6, 11 y 12 se sembraron productos amplificados de 621 pb obtenidos mediante cebadores LCR2.
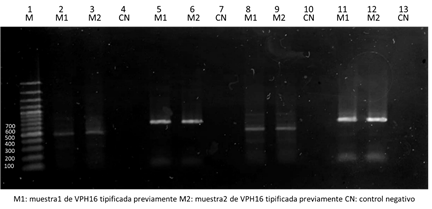
Figura 3. Corrida electroforética en gel de agarosa al 1% de productos amplificados de VPH 16 con cebadores LCR1 y LCR2 utilizando una temperatura de anillamiento de 60ºC. Carril 1: Marcador de pares de base. Carriles 2-7 se utilizó buffer HF y del 8-13 buffer GC. En los carriles 2, 3, 8 y 9 se sembraron productos amplificados de 460 pb, obtenidos mediante cebadores LCR1. En los carriles 5, 6, 11 y 12 se sembraron productos.
Optimización de la PCR E6
Para la PCR E6, se observó una mejor amplificación utilizando una temperatura de anillamiento de 50°C con el buffer GC. En la Figura 4 se observa la corrida electroforética de los productos amplificados con los cebadores E6, a diferentes temperaturas de anillamiento y con diferentes buffers de reacción.
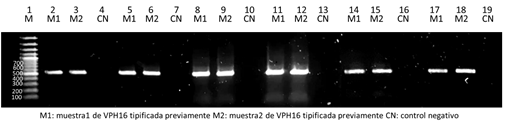
Figura 4. Corrida electroforética en gel de agarosa al 1% de productos amplificados de VPH 16 con cebadores E6, con un producto de amplificación de 524pb. En los carriles del 2-7 se sembraron los productos de amplificación obtenidos utilizando una temperatura de anillamiento de 48ºC, del 8 al 13 de 50ºC y del 14-19 de 52ºC. En los carriles 2, 3, 4, 8, 9, 10, 14, 15 y 16 se sembraron productos obtenidos con la utilización del buffer HF y en los carriles 5, 6, 7, 11, 12, 13, 17, 18 y 19 buffer GC.
Por tanto, para la amplificación de la región LCR utilizando los cebadores LCR1 y LCR2 se observaron mejores resultados a una temperatura de anillamiento de 57°C y para la amplificación del gen E6 50°C. La concentración de MgCl2 utilizada en ambas reacciones fue de 1,5mM, la de dNTP 0,2mM y la de cebadores 0,2uM.
DISCUSIÓN
Para ambas reacciones de PCR se optó por utilizar el buffer GC, ya que se obtuvieron bandas más nítidas para LCR y para E6. Estos resultados se correlacionan con las indicaciones establecidas por el fabricante de la enzima ADN polimerasa utilizada, iProof High-Fidelity DNA Polymerase de la marca Bio-Rad, donde se recomienda la utilización del buffer GC para la amplificación de templados ricos en bases G-C y para secuencias complejas o de mayor longitud16.
Las temperaturas de anillamiento testadas para la amplificación de la región LCR y el gen E6 fueron seleccionadas teniendo en cuenta la temperatura de melting (Tm) de cada par de cebadores, así como también las indicaciones proveídas por el fabricante de la enzima utilizada, el cual sugiere la elección de una temperatura de anillamiento que se encuentre 3°C por encima de la Tm más baja del par de cebadores utilizados16. Las temperaturas de anillamiento seleccionadas para cada reacción de PCR permitieron la obtención de bandas nítidas del tamaño esperado para cada producto de amplificación, además de minimizar la amplificación de fragmentos inespecíficos. Sin embargo, cabe mencionar que en algunos casos se observaron productos inespecíficos, los cuales fueron muy tenues en comparación al producto deseado, lo cual permitirá realizar una purificación en gel a fin de secuenciar el producto específico.
Para los ensayos de amplificación de la región LCR se optó por la temperatura de anillamiento de 57°C, lo cual permitió realizar las reacciones de amplificación de los segmentos LCR 1 y LCR 2 bajo las mismas condiciones de ciclado, con Tm respectivos de 59°C y 53°C17. Esta temperatura seleccionada es similar a la utilizada en los experimentos de Xi et al.13.
Para la amplificación del segmento E6 se optó por la temperatura de anillamiento de 50°C, lo cual se concuerda con una Tm calculada de 53°C17, similar a lo reportado en los ensayos realizados por Cornet et al.14.
En conclusión, se logró optimizar las reacciones para la amplificación de la región LCR y del gen E6 del genoma de VPH16, obteniéndose amplicones con concentraciones de ADN necesarias para ser sometidos a secuenciación por el método de Sanger, según inspección visual de los productos obtenidos en geles, mediante la comparación con las bandas del marcador de peso molecular, cuya concentración es conocida. La presente optimización servirá para determinar las variantes de VPH 16 mediante el análisis de las secuencias de ambas regiones. Además, los amplicones podrán ser clonados y utilizados en ensayos in vitro de funcionalidad y actividad transcripcional de la región no codificante LCR, considerando que han sido reportadas diferencias en la capacidad de patogénesis cervical de los diferentes linajes de VPH 16.

