











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
Permalink
Introducción
La yerba mate es una especie vegetal denominada Ilex paraguariensis A. St.-Hil. (Grigioni et al., 2004), reconocida por el botánico francés Auguste François César Prouvençal de Saint-Hilaire (Gugliucci, 1996). Pertenece a la clase de las dicotiledóneas, dialipétalas corolianas, familia Aquifoliáceae, del género Ilex que comprende casi toda la familia (175 de las 181 especies) que se encuentran dispersas en toda Sudamérica. Sus hojas son alternas, coriáceas, de forma obovada, elípticas, con borde aserrado, resoluto. Sus dimensiones difieren entre 5 y 15 centímetros (cm) de largo por 2 a 5 de ancho que perduran en la planta unos tres años.
En estado de plena madurez las hojas son espesas, duras y lucen como enceradas (Fig. 1, (a)), de color verde más intenso en su cara superior que en la inferior presentando un corto pecíolo de color claro verdoso, a veces ligeramente rosado. La floración tiene lugar entre los meses de octubre a diciembre, es de tipo racimosa, en forma de falsas panojas, desarrollándose en las axilas de las hojas y en la base de las ramitas en número de 40 a 50 flores por racimo. Sus flores son pequeñas, dioicas, con cáliz y corola de constitución tetrámera. La raíz de color marrón es de tipo pivotante, con raíces secundarias que se insinúan en el mismo sentido (Zelada Cardozo & González Villalba, 2019).

El proceso de elaboración de la yerba mate comercial puede dividirse en dos grandes etapas: la primera que va desde la cosecha de yerba mate verde, que llega hasta la obtención la yerba mate canchada y estacionada y la otra que se inicia en el molino llegando a las distintas presentaciones de los productos para consumir de diferentes maneras (Arbiser, J.L. et al., 2005; Gugliucci, 1996). La yerba mate comercial (Fig. 1b) se utiliza para hacer una bebida refrescante cuando hace calor, denominada tereré que se sirve con agua fría. Servida con agua caliente la bebida es conocida como mate (Arcari et al., 2009; Bastos et al., 2006). Otra bebida muy consumida en nuestro país y producida a base de yerba mate es el mate cocido que es una infusión típica de la gastronomía. Por su valor nutricional y energizante propio, y por brindar sensación de saciedad se lo suele beber en el desayuno o me- rienda, acompañado con un alimento sólido como pan u otros productos de confitería. Estas bebidas mencionadas se consumen tradicionalmente en las regiones centro y sur de Sudamérica, principalmente en Paraguay, Argentina, Uruguay, sur y centro oeste de Brasil, la región del Chaco de Bolivia y el sur de Chile (Barchuk R.D. et al., 1998; Zelada Cardozo & González Villalba, 2019).
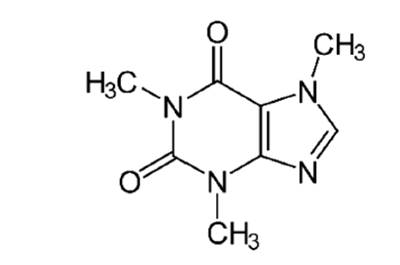
Unos de los componentes más importantes que contiene la yerba mate es la cafeína (Fig. 2) que es el responsable de la acción estimulante de estas bebidas, los taninos le dan el sabor astringente, y la espuma producida al cebar principalmente en el mate es debida a las saponinas triterpénicas (matesaponinas) (Cardozo et al., 2007; Isolabella et al., 2010). La cafeína es un alcaloide del grupo de las xantinas, que actúa como droga estimulante y psicoactiva. Su fórmula química es C8H10N4O2, con una masa molecular de 194,19 g/mol. Es una molécula química aquiral, y, por lo tanto, no tiene enantiómeros ni tiene estereoisómeros (Bertoni et al., 1992; Bracesco et al., 2003).
Su nombre sistemático es 1, 3,7-trimetilxantina, 1, 3,7-trimetil-2,6-dioxopurina o 3,7-dihidro-1, 3,7- trimetil-1H-purina-2,6-diona. También es conocida como trimetilxantina, teína, mateína, guaranína, metilteobromina o metilteofilina, ya que también se obtiene por extracción de materiales vegetales como el café, té, guaraná, chocolate, yerba mate o la nuez de cola (Clifford et al., 1990; Heck & Mejía, 2007; Carini et al., 1998).
En esta investigación se ha desarrollado y validado dos técnicas analíticas para la determina- ción y cuantificación de la cafeína en yerba mate comercial, indicando la cantidad de cafeína que contiene la yerba mate por kilogramo, utilizando como medida de cantidad una guampa, que es el recipiente tradicional para el consumo del mate y tereré. En esta guampa se agregó la yerba mate a analizar, se pesó la muestra y a partir de ahí se desarrolló la técnica de extracción. En este trabajo se pretende conocer cuanta cafeína se puede extraer utilizando la técnica de extracción sólido - líquido a reflujo con un extractor de Soxhlet y posterior separación del analito de la fase acuosa por extracción líquido-líquido.
Materiales y métodos
Productos químicos y reactivos
Se han utilizados reactivos de grado analítico. El patrón de cafeína fue suministrado por Sigma-Aldrich (St. Louis, MO). La solución madre del analito fue preparada una sola vez a una concentración de 400 µg. mL-1 y se almacenó a -20 °C en botella de vidrio oscuro. El acetonitrilo que se utilizó para la preparación de la fase móvil fue de grado cromatográfico (Merck, Darmstadt, Alemania), al igual que el ácido clorhídrico y el cloroformo utilizadas en la extracción del analito. Los reactivos carbonato de sodio y sulfato de sodio empleados fueron de calidad analítica (Merck, Darmstadt, Alemania). El agua destilada se obtuvo en un equipo (QUIMIS, Brasil) y posteriormente purificada en un equipo Barnstead ™ MicroPure ™.
Instrumentación y Software
Para la extracción de la cafeína de la matriz estudiada se utilizó un equipo de extracción Soxhlet, un embudo de decantación para la separación líquido- líquido y una variedad de materiales de vidriería. Para su cuantificación se empleó primeramente un espectrofotómetro Thermo ScientificTM UV-Visible GenesysTM 10S equipado con una lámpara de Xenón de alta intensidad y una geometría óptica de doble haz utilizando un software VISIONlite™ (versión 5.0).
Las muestras se colocaron en cubetas de cuarzo de 1 mm de paso óptico y 2,5 mL de volumen útil con adaptador para porta cubetas. Posteriormente los ensayos se realizaron en un cromatógrafo de líquidos de alta resolución HPLC Shimadzu (Japón) modelo DGU-20A5 E con una bomba cuaternaria, inyector automático y detector de fotodiodos PDA y/o de fluorescencia FLD. La separación cromatográfica del analito se obtuvo utilizando una columna ZORBAX Eclipse XDB-C18 (150 mm x 4.6 mm; 5 µm).
El control del instrumento, la detección del pico y la integración del mismo se llevaron a cabo utilizando el software LCsolution.exe (versión 1.0) (Shimadzu, Japón). Para las pesadas de los reactivos y las muestras se empleó una balanza analítica RADWAG 310.R2 (Polonia). Para el tratamiento estadístico de los datos se utilizó Statgraphics Plus versión 5.0 (Manugistics, Rockville, MD, EE. UU., 2000) y Microsoft® Office 2010.
Preparación de las soluciones patrones
Primeramente, se ha preparado una disolución patrón de cafeína, a partir de 40 mg de cafeína previamente secada que se disolvieron en un matraz aforado de 100 mL en agua destilada (400 µg. mL-1). Seguidamente se traspasaron con ayuda de una pipeta aforada 10 mL de esta disolución a otro matraz aforado de 100 mL y se llevó a enrase con agua destilada (40 µg. mL-1). A partir de esta última disolución patrón de 40 µg. mL-1, se prepararon 5 disoluciones más diluidas de concentraciones de 4, 8, 16, 24, y 32 µg de cafeína por cada mL de solución:
(5 mL de solución patrón de cafeína de 40 µg. mL-1 + 2 mL de HCl 0,01 M) en 50 mL con agua destilada (4 µg. mL-1).
(5 mL de solución patrón de cafeína de 40 µg. mL-1 + 1 mL 0,01 M) en 25 mL con agua destilada (8 µg. mL-1).
(10 ml de solución patrón de cafeína de 40 µg. mL-1 + 1 mL HCl 0,01 M) en 25 ml con agua destilada (16 µg. mL-1).
(15 ml de solución patrón de cafeína de 40 µg. mL-1 + 1 mL HCl 0,01 M) en 25 ml de agua destilada (24 µg. mL-1).
(20 ml de solución patrón de cafeína de 40 µg. mL-1 + 1 mL HCl 0,01 M) en 25 ml de agua destilada (32 µg. mL-1).
Extracción de la muestra
Las muestras de yerba mate (Ilex paraguariensis) fueron adquiridas de un centro comercial de la ciudad de Asunción, dichas muestras fueron de un producto tradicional sin aditivos. Se analizaron por triplicado; se pesaron 45 gramos de yerba mate comercial que es el peso aproximado que contiene una guampa promedio de tereré (se utilizó una guampa para medir el peso aproximado y por triplicado), y se llevó a reflujo con 400 mL de agua durante 15 minutos en un equipo de reflujo Soxhlet. Seguidamente, se filtró la solución en caliente y se añadió 5 gramos de Na2CO3 hasta la total disolución. Se dejó enfriar y se agregaron 30 mL de cloroformo que se extrajeron agitando suavemente durante 3 a 5 minutos en un embudo de decantación y se repitió la operación una vez más.
Después de la separación de las dos fases, se añadieron pequeñas cantidades de Na2SO4 para absorber el agua, se filtró la fase clorofórmica haciéndola pasar por lana de vidrio y posteriormente por filtración al vacío con papel de filtro en un embudo de Büchner, el filtrado se colocó en un vaso de precipitado y se dejó evaporar hasta sequedad. Una vez eliminado el cloroformo, se añadió al mismo vaso 50 mL de agua destilada y se agitó bien hasta disolver totalmente. Posteriormente se trasvaso el líquido disuelto a un matraz aforado de 100 mL y se llevó a enrase. Finalmente, se tomaron 1 mL de esta última solución, se añadió 4 mL de la solución de ácido clorhídrico 0,01 M y se enrasó a 100 mL con agua destilada. Para los ensayos en el UV-visible las muestras y patrones fueron utilizadas de manera directa mientras que para el HPLC fueron filtradas previamente por filtro de membrana de Nylon (Poliamida), Whatman, 0.45 µm utilizando una jeringa.
Medidas espectroscópicas, UV-Visible
Primeramente, se realizó un barrido espectral de 200 a 400 nm teniendo en cuenta las características químicas de la molécula, con una solución patrón de 32 µg. mL-1 de cafeína, descrita su preparación en el apartado de Preparación de las soluciones patrones, y una vez obtenida la longitud de onda de absorbancia máxima para el analito se procedió a la validación y su cuantificación. En todo momento se empleó un blanco de disolvente para realizar la corrección de la línea de base.
Condiciones cromatográficas, HPLC
Las condiciones cromatográficas del HPLC fueron optimizadas. Para la optimización del método cromatográfico se empleó una disolución acuosa que contenía la cafeína a una concentración de 32 μg L-1. Dadas las características del analito, en el desarrollo de la metodología propuesta se ha empleado la cromatografía en fase reversa (fase estacionaria menos polar que la fase móvil). Se seleccionó de acuerdo a las características fisicoquímicas del analito una columna con relleno C18 ZORBAX Eclipse XDB (150 mm x 4.6 mm; 5 µm) de dimensiones y tamaño de partícula. Se estudiaron las principales variables que afectan a la separación cromatográfica y la intensidad de la señal, se evaluaron diferentes fases móviles de acuerdo a la naturaleza del analito, se ensayaron la mezcla Agua: Acetonitrilo a diferentes proporciones (70:30), (80:20) y (90:10). También se evaluó el efecto del caudal, la temperatura de la columna y volúmenes de inyección. Se ensayaron velocidades de flujo de 0,5 a 1,5 mL.min-1. Se en- sayaron temperaturas de la columna de 30 °C a 50 °C. Finalmente se evaluó el volumen de inyección desde 5 a 30 µL.
Validación de los métodos analíticos de cuanti ficación
Los parámetros de validación evaluados fueron: linealidad, límites de detección y cuantificación, rango dinámico lineal, sensibilidad analítica y exactitud del método expresada en términos de precisión y veracidad (USP 29, 2005). Los requisitos de la validación del método analítico se establecieron de la siguiente manera: (a) Linealidad, el coeficiente de determinación (R2) debe ser igual o mayor que 0,999 y la desviación residual máxima debe ser menor al 25%. (b) La precisión, expresada como desviación estándar relativa DER (precisión entre días) debe ser ≤ 30%. (c), la veracidad, expresada como recuperación media, debe estar en el rango 70 a 120%. (d) El límite de cuantificación (LC) debe cumplir con los requisitos (b) y (c). Estos requisitos se ajustan a la Decisión 2002/657 / CE de la Comisión de la Comunidad Europea (14).
Linealidad
Para el establecimiento de esta función de calibra do, se prepararon patrones conteniendo cantidades crecientes del analito correspondiente 4, 8, 16, 24, 32 µg. mL-1 (3 réplicas por cada nivel). Se realizó la estimación de la ecuación de la recta, bajo condiciones de reproducibilidad y se determinó el valor del coeficiente de correlación lineal que considera un criterio de aceptabilidad igual o superior a r ≥ 0,999. La ecuación que se utilizó para determinar la linealidad del método es la siguiente, y = mx+b, dónde y = Área que representa la señal analítica; x = concentración del estándar de la cafeína (µg. mL-1), m = pendiente de la recta, b = ordenada al origen (Analytical Methods Committee, 1994; IUPAC, 1978; Dean & Dixon, 1951; Blanco & Cela, 1994).
Límites de detección y cuantificación
El límite de detección (LD) es la mínima concentración detectable de manera confiable por la técnica. Para la determinación de dicho parámetro se ha calculado en función de la desviación estándar de la concentración predicha para una muestra en blanco (So) (Currie, 1999). Primeramente, para estimar So se recurrió a la ecuación escrita del modo siguiente:
y partir de ahí se calculó el límite de detección que es el resultado de la multiplicación de So por 3.
Mientras que el límite de cuantificación (LC) es la mínima concentración cuantificable en forma confiable. Que se tomó como la concentración correspondiente a 10 veces la desviación estándar (en unidades de concentración) del blanco (So por 10). (González et al., 1996; Cuadros Rodríguez et al., 1993)
Rango Dinámico Lineal.
El Rango Dinámico Lineal (RDL) fue establecida como el intervalo de concentraciones que compren- de entre el límite de cuantificación de los métodos y el límite superior del intervalo de concentraciones en el que se ha aplicado a los métodos estudiados (IUPAC, 1978).
Sensibilidad Analítica.
La sensibilidad de un método analítico o sensibilidad de la calibración mide la relación entre la señal instrumental y la concentración del analito, y viene dada por la pendiente de la recta de calibración (González et al., 1996). Para este trabajo de investigación se propuso que la sensibilidad analítica (SAnalitica) está definida por el cociente entre la desviación estándar de la regresión (Sy/x) y la pendiente del calibrado (b), según la ecuación SAnalítica = Sy/x / b (Mandel & Stiehler, 1954).
Exactitud del y Método: Precisión Veracidad.
Para establecer la exactitud de los métodos propuestos, se han estudiado la veracidad del mismo en términos de recuperación con muestras dopadas y su precisión en términos de variabilidad inter e intra-día. Para el estudio de exactitud del método, en términos de veracidad , se ha llevado a cabo un estudio de recuperación a tres niveles de concentración (16, 24, 32 µg. mL-1 ).
Las muestras fueron analizadas empleando el procedimiento operatorio descrito a lo largo del artículo y la concentración en la muestra se determinó por interpolación a la curva de calibración dentro del rango dinámico lineal. Los valores de recuperación se obtuvieron por comparación con las cantidades de analito conocidas que fueron añadidas previamente a las muestras en blanco (los disolventes utilizados para la extracción durante el reflujo fueron dopadas con las concentraciones mencionadas y se realizó todo el procedimiento de extracción) (Benítez-Villalba et al, 2018)
Para evaluar la exactitud del método en términos de precisión , se estudió la repetibilidad (precisión “intra-día”) y la reproducibilidad (precisión “inter-día”). Para ello, se realizaron diversas réplicas de los análisis llevados a cabo en el estudio de veracidad, durante un mismo día y durante tres días consecutivos. Cada muestra dopada en blanco fue extraída y analizada por triplicada, es decir tres réplicas de cada punto en el mismo día para evaluar la variabilidad “intra-día” y se repitió en tres días consecutivos para evaluar la precisión, expresada como desviación estándar relativa (DER) (ISO/TC 69/SC 6, 1994a & 1994b); Commission of the Eu ropean Communities, 2002; Massart et al., 1997).
Aplicación de los métodos analíticos (identificación y cuantificación)
Una vez finalizada la validación de los métodos analíticos se procedió a la determinación y cuantificación de la cafeína en las muestras de yerba mate utilizando la curva de calibrado descrito en la metodología para las dos técnicas analíticas en estudio, el espectrofotométrico y cromatográfico. Primeramente, las muestras fueron cuantificadas con la técnica espectrofotométrica (UV-visible). Subsiguientemente se han cuantificadas las mismas muestras en el cromatógrafo líquido de alta resolución (HPLC) y posteriormente se promediaron los resultados obtenidos por ambas técnicas.
Resultados y discusión
UV-Visible
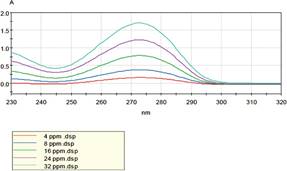
Para el método espectrofotométrico UV-visible se determinó la longitud de onda de máxima absorción del analito que fue de 273 nm. Este valor de la longitud de onda también fue utilizado para la determinación y cuantificación de cafeína por HPLC. En la Fig. 3 se representan los espectros de absorción de las soluciones patrones de cafeína en agua destilada.
HPLC
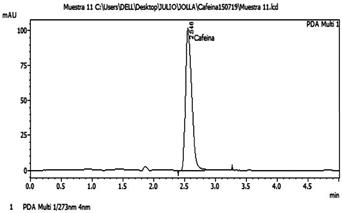
De las condiciones cromatográficas que fueron optimizadas en el HPLC la fase móvil tuvo un óptimo que fue el Agua: Acetonitrilo en una proporción (80:20) por dar una mejor señal analítica; el óptimo para la velocidad de flujo fue de 1 mL. min -1 que mejoró significativamente la resolución, la forma del pico, la intensidad de respuesta y el tiempo de retención; la temperatura óptima de la columna fue de 30 °C que mostró una buena forma de pico y el volumen de inyección óptimo fue de 20 µL cuyos resultados en el pico del cromatograma de una muestra de yerba mate comercial se puede observar en la Fig. 4.
de calibración, b = pendiente, Sb = desviación están dar de la pendiente, a = intercepto, Sa = desviación estándar del intercepto, Sy/x = desviación estándar de la regresión, % plof = valor P de prueba de falta de ajuste, LD = límite de detección, LC = Limite de cuantificación, RDL = rango dinámico lineal (Mandel & Stiehler, 1954).
A continuación, se detallan los resultados obtenidos Tabla 1.
Validación de métodos analíticos
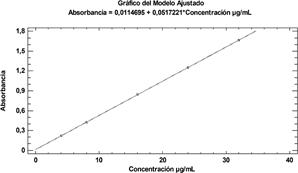
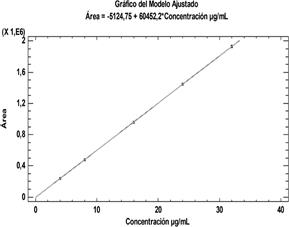
Linealidad: para ambas técnicas analíticas se ha demostrado ser significativos con R2= 99,999%. Tanto los interceptos como las pendientes son significativamente diferentes de cero. Las gráficas de linealidad de los valores observados y ajustados por mínimos cuadrados se muestran en la Fig. 5 (UV-Visible) y Fig. 6 (HPLC). Estos métodos analíticos de acuerdo a los resultados obtenidos han demostrados ser altamente sensibles en la detección de la cafeína, con límites de detección (LD) de 0,101 µg. mL-1 para la técnica espectrofotométrica (UV-Visible) y 0,092 µg. mL-1 para la técnica cromatográfica HPLC; mientras que los límites de cuantificación (LC) para ambas técnicas fue de 0,307 µg. mL-1.
Tabla 1. Parámetros de calibración en las técnicas analíticas espectrofotométrico y cromatográfico.
| Linealidad | ||
|---|---|---|
| Parámetros | Espectrofotométrico | Cromatográfico |
| Ecuación de la recta | Y = 0,051721*X + 0,0114695 | Y= 60452,2*X - 5124,75 |
| R2 (%) | 99,999 | 99,999 |
| n | 15 | 15 |
| b (µg. mL-1 ) | 0,052 | 60452,2 |
| sb | 4,6E-05 | 48,471 |
| a | 0,011 | 5124,750 |
| sa | 0,001 | 953,773 |
| sy/x | 0,002 | 1923,250 |
| % plof | 0,000 | 0,124 |
| LD (µg. mL-1 ) | 0,101 | 0,092 |
| LC (µg. mL-1 ) | 0,307 | 0,307 |
| Sensibilidad | 0,023 | 0,032 |
| RDL (µg. mL-1 ) | 0,307 - 32 | 0,307 - 32 |
El Rango Dinámico Lineal (RDL): para ambas técnicas analíticas el RDL están comprendidas entre el límite de cuantificación 0,307 µg. mL-1 y el límite superior de la concentración que se aplicó a los dos métodos 32 µg. mL-1.
Sensibilidad Analítica: La sensibilidad del método analítico de la calibración que se ha calculado a partir de la regresión lineal para ambos métodos analíticos, fueron respectivamente de 0,023 para UV-visible y 0,032 para el HPLC.
Exactitud del Método: Los estudios de recuperación se realizaron para las dos técnicas analíticas validadas, previa extracción del analito en el disolvente fortificada a tres niveles de concentración (16, 24, 32 µg. mL-1), obteniendo para la técnica analítica de UV-Visible recuperaciones de inter e intra día entre 80% y 84%, mientras que para la técnica por HPLC las recuperaciones inter e intra día se encontraron entre 81 y 84%, lo que nos lleva a deducir que el procedimiento de extracción es eficiente y el método veraz. Por otro lado, los valores obtenidos en ambas técnicas analítica la DER fueron menor 1% en todos los casos.
Estos datos se encuentran dentro de límites aceptables para cumplir los requisitos establecidos en la guía de validación empleada, según la cual se consideran aceptables valores iguales o inferiores al 15 % en general, y al 20 % en la zona próxima al límite de detección del método, por lo que se puede concluir que el método propuesto de la técnica de extracción a reflujo con soxhlet y posterior separación del analito por líquido-líquido genera resultados próximos entre sí, cumpliendo de este modo los requisitos de precisión (Cuadros Rodríguez et al., 1993). Se trata por tanto de un método veraz, preciso y por tanto exacto. En las Tablas 2 y 3 se muestran los valores de recuperación obtenidos para ambas técnicas analíticas
Tabla 2. Ensayos de recuperación para la determinación de la exactitud en términos de veracidad del método utilizando la técnica de extracción liquido-liquido.
| Ensayo Intra-dia | ||||||
|---|---|---|---|---|---|---|
| Analito Cafeína | Dopado µg. mL-1 | Observado µg. mL-1 | DE | Recuperación % | DER % | n |
| UV-Visible | 16 | 13,0028 | 0,0193 | 81,2675 | 0,1487 | 3 |
| 24 | 19,5120 | 0,0112 | 81,2998 | 0,0572 | 3 | |
| 32 | 25,6344 | 0,0403 | 80,1076 | 0,1570 | 3 | |
| 16 | 13,1418 | 0,0057 | 82,1365 | 0,0435 | 3 | |
| HPLC | 24 | 20,2089 | 0,0076 | 84,2038 | 0,0375 | 3 |
| 32 | 26,0293 | 0,0685 | 81,3414 | 0,2629 | 3 | |
Tabla 3. Ensayos de recuperación para la determinación de la exactitud en precisión de veracidad del método utilizando la técnica de extracción liquido-liquido.
| Ensayo Inter-día | ||||||
|---|---|---|---|---|---|---|
| Analito Cafeína | Dopado µg. mL-1 | Observado µg. mL-1 | DE | Recuperación % | DER % | n |
| UV-Visible | 16 | 13,1575 | 0,0199 | 82,2342 | 0,1469 | 3 |
| 24 | 20,2402 | 0,0914 | 84,3342 | 0,4514 | 3 | |
| 32 | 26,3859 | 0,4514 | 82,4559 | 0,7154 | 3 | |
| 16 | 13,2301 | 0,0089 | 82,6879 | 0,0673 | 3 | |
| HPLC | 24 | 19,4921 | 0,0945 | 81,2171 | 0,4848 | 3 |
| 32 | 26,6358 | 0,0278 | 83,2369 | 0,1044 | 3 | |
Aplicación de los métodos analíticos a muestras reales
Finalizado el desarrollo y validación de los métodos analíticos se procedió a la determinación y cuantificación de la cafeína en la extracción de las muestras de yerba mate que fueron procesadas, utilizando la curva de calibrado descrito en la metodología para ambas técnicas analíticas.
Los resultados obtenidos de la extracción de cafeína en yerba mate analizada para la técnica analítica por espectrofotometría (UV-visible) fue de 127,6 mg/45g de muestra mientras que para la técnica cromatográfica (HPLC) se ha obtenido como resultado 121,2 mg/45g de muestra promediando así los resultados para ambas técnicas de 124,4 mg de cafeína por cada 45 gramos de yerba mate. En la Tabla 4 podemos observar los resultados obtenidos.
Conclusión
Este trabajo de investigación propone la validación de dos metodologías analíticas para la determina- ción y cuantificación de la cafeína en muestras de yerba mate comercial, la espectroscopia UV-visible y la cromatografía de líquidos HPLC, para ello se ha utilizado la técnica de extracción sólido-líquido a reflujo con Soxhlet y posterior separación de las fases acuosas por extracción líquido-líquido. Esta técnica de extracción podemos considerar una técnica de extracción optima debido a un buen rendimiento de extracción obtenido y de fácil operación. La cafeína es el componente más importante que contiene está matriz que es el responsable de la acción estimulante en las bebidas que son preparadas a base de ella. Las técnicas analíticas UV-visible y el HPLC empleadas para el análisis de los extractos obtenidos a partir de las muestras mostraron una alta sensibilidad analítica y precisión, tiempos de análisis corto y el alto grado de automatización. Se ha podido estimar que en 45 gramos de yerba mate comercial contiene aproximadamente 124,4 mg de cafeína.
Tabla 4. Determinación de la concentración de la cafeína en muestras de yerba mate en las técnicas analíticas UV-visible y por HPLC.
| Concentración de la cafeína en muestras de yerba mate mg/45 g de muestra | |||||
|---|---|---|---|---|---|
| Analito | UV-Visible | Promedio UV-Visible | HPLC | Promedio HPLC | Promedio ambos métodos |
| 127,9 | 118,1 | ||||
| Cafeína | 126,6 | 127,6 | 125,7 | 121,2 | 124,4 |
| 129,3 | 119,8 | ||||
La contribución de este trabajo de investigación es el aporte de las metodologías analíticas que ofrecen una importante innovación científica, ya que en la actualidad no existe ningún trabajo publicado sobre el análisis de este analito en esta matriz en nuestro país. Estas técnicas validadas podrán ser utilizada en futuras investigaciones en otras matrices que contengan este analito.

