Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los virus de la influenza (VI) sufren continuamente cambios genéticos, ya sea mediante sustituciones puntuales de aminoácidos (aas) denominados deriva antigénica, o por intercambio de segmentos completos, para evadir la presión del sistema inmune del huésped y adaptarse a nuevos huéspedes. En el año 2009, la circulación de una cepa triple reasortante de Influenza A (A/H1N1pdm2009), desplazó a las previamente circulantes, y ha estado evolucionando continuamente desde entonces hasta la actualidad2-4. Los cambios experimentados en los aas del virus pueden alterar sus características antigénicas, su virulencia y su sensibilidad a los antivirales5.
La infección por Influenza A(H1N1)pdm09 es principalmente una enfermedad del tracto respiratorio superior leve y autolimitante. El espectro de presentación clínica varía desde casos asintomáticos hasta neumonía viral primaria que resulta en dificultad respiratoria aguda, falla multiorgánica y muerte6. La patogenicidad del virus de la influenza depende de la función de las proteínas virales y de la respuesta inmune del huésped7, de modo que tanto factores virales como del huésped inciden en la patogénesis de la gripe.
El análisis filogenético basado en los cambios de nucleótidos que conducen a sustituciones de aas en las proteínas de superficie Hemaglutinina (HA) y Neuraminidasa (NA) del VI, en conjunto con los análisis antigénicos, son realizados como parte de la vigilancia virológica, a fin de monitorear el impacto en la severidad de los casos y realizar recomendaciones apropiadas sobre las cepas a ser incluidas en las vacunas anuales8. Desde su primera detección en Paraguay, en junio de 2009, el VI A(H1N1)pdm09 ha circulado en el país todas las temporadas siguientes, ocasionando tanto casos de enfermedad tipo influenza (ETI) en pacientes ambulatorios como Infecciones respiratorias agudas graves (IRAG) en hospitalizados.
El objetivo de este trabajo es estudiar la diversidad genética del segmento HA de los virus influenza A (H1N1) 2009 circulantes en Paraguay entre 2009 y 2016.
Materiales y métodos
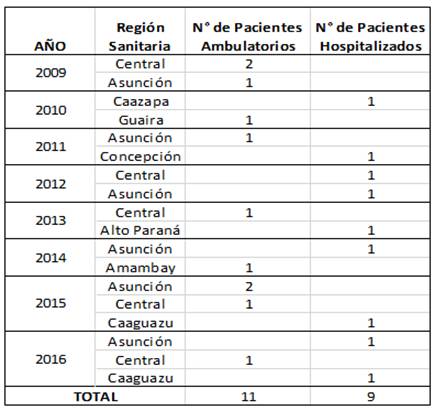
Se estudiaron muestras representativas de pacientes provenientes de diferentes regiones del Paraguay, captados por la Vigilancia Centinela de ETI e IRAG entre los años 2009 y 2016. Fueron seleccionados 20 pacientes con diagnóstico confirmado de VI A(H1N1)pdm09, 11 de ellos ambulatorios con ETI y 9 hospitalizados con IRAG, a fin de investigar diferencias en la secuencia genómica de los virus aislados a partir de sus muestras. Los datos clínicos y demográficos de los pacientes fueron obtenidos de las fichas epidemiológicas completadas en los centros asistenciales.
Fueron analizadas las secuencias nucleotídicas del Gen HA de las 20 cepas de VI A(H1N1)pdm09, detectadas por RT-PCR en Tiempo Real y aisladas en el Centro Nacional de Influenza (CNI) de Paraguay entre los años 2009 a 2016; y posteriormente secuenciadas en el Centro Colaborador de OPS/OMS en Atlanta USA (CDC). Todas las secuencias, incluidas las de referencia fueron tomadas de la base de datos “Global Initiative on Sharing All Influenza Data” (GISAID EpiFlu™). Las secuencias fueron alineadas con cepas de referencia y editadas mediante el programa Bioedit y la aplicación Muscle. El análisis filogenético fue realizado por el método neighbor-joining utilizando el programa MEGA 6. Las sustituciones de aminoácidos y variaciones en los sitios de glicosilación fueron tabuladas utilizando la aplicación Cubit, comparando con el virus vacunal A/California/07/2009 (H1N1).
Resultados
Los datos de procedencia y hospitalización de los pacientes estudiados se describen en la Tabla 1.
El análisis filogenético muestra la circulación de cepas pertenecientes al menos a 5 grupos genéticos (5, 6, 6B, 6C, 6B.1) de VI A(H1N1)pdm09 en Paraguay desde el 2009 (Figura 1).
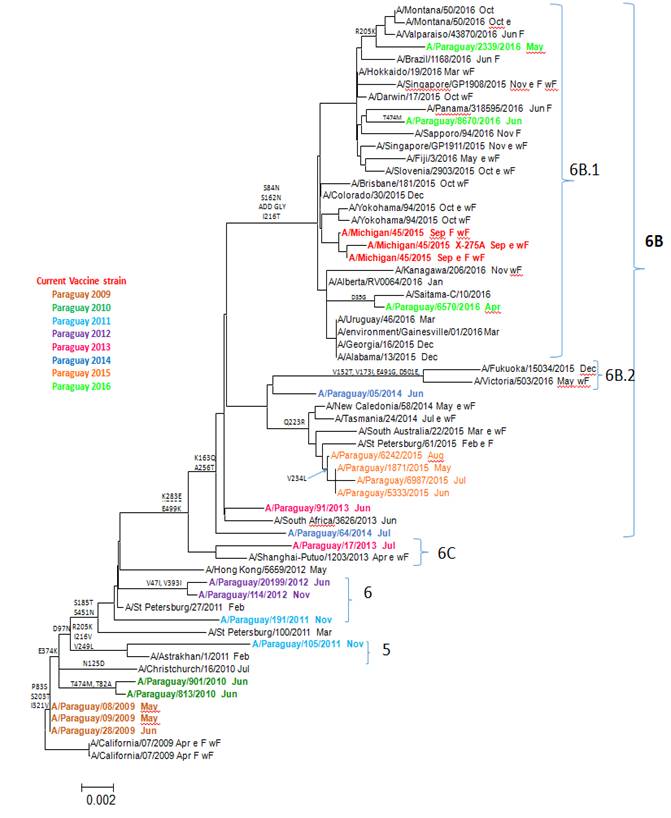
Figura 1. Relaciones evolutivas entre la gripe A (H1N1) pdm09 Gen de hemaglutinina (HA). Paraguay 2009-2016 virus del 2009 fueron muy cercanos al virus de referencia A/California/07/2009 (H1N1), con solo 3 sustituciones de aas (P83S, S203T, I132V). Se nota una tendencia a incrementar progresivamente la frecuencia del número acumulado de sustituciones de aas las siguientes temporadas de influenza, con 6 y 8 sustituciones en el 2010, entre las que destacan como nuevas: E374K, T474M, T82A (Tabla 2).
Tabla 2. Sustituciones de aminoácidos en el segmento HA de los virus Influenza A(H1N1) pdm09 circulantes en Paraguay 2009-2016
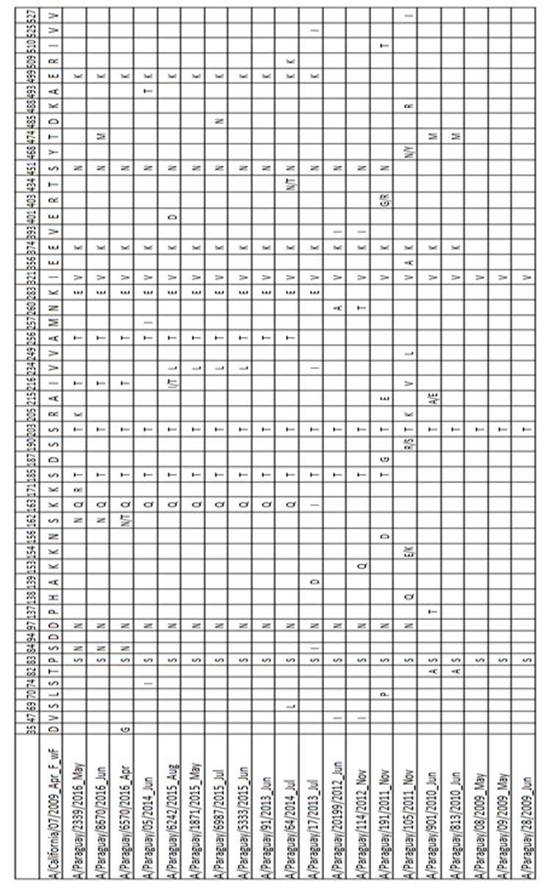
Una de las cepas del 2011 pertenece al Grupo 5, caracterizado por las sustituciones R205K, I216V, V249L. Otra cepa del 2011 y las del 2012 pertenecen al Grupo 6 caracterizado por las sustituciones S185T y S451N, además todos presentan la mutación D97N. Las cepas del 2012 presentan las sustituciones adicionales V47I y V393I. Uno de los virus aislados en 2013 pertenece al grupo 6C caracterizado por las sustituciones de aas K283E y E499K; mientras que otro virus del 2013 al igual que los del 2014 y 2015 quedaron en el Grupo 6B caracterizado por las sustituciones K163Q y A256T. Solamente los virus aislados en 2016 pertenecen al sub Grupo genético 6B.1 con la característica sustitución S162N que agrega un motivo de glicosilación (Tabla 3), además de las sustituciones en S84N y I216T. En este mismo grupo se encuentra la actual cepa vacunal A/Michigan/45/2015.
Tabla 3. Cambios en los Sitios de glicosilación del segmento HA de los virus Influenza A(H1N1)pdm09 circulantes en Paraguay 2009-2016
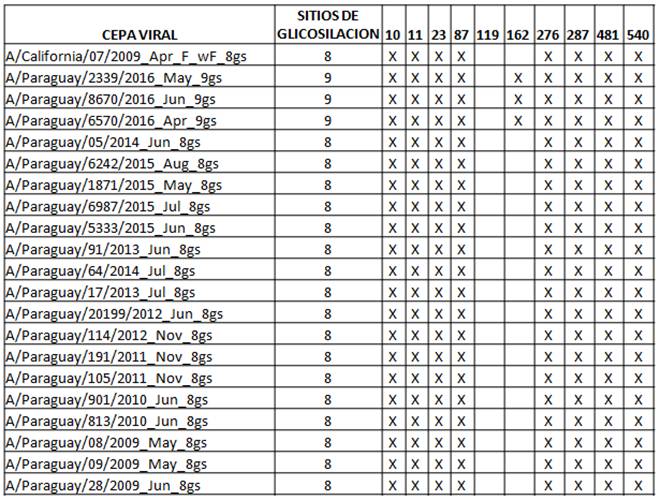
No se encontraron diferencias resaltantes entre las secuencias de lo virus pertenecientes a pacientes ambulatorios y hospitalizados dentro de cada año. Tampoco se detectaron diferencias importantes según región geográfica dentro del mismo periodo de tiempo.
Discusión
En el presente estudio hemos identificado al menos 5 grupos genéticos diferentes de VI A(H1N1)pdm09 que circularon en Paraguay desde su primera introducción en junio del 2009 hasta el año 2016. Esta agrupación está basada en variaciones de aas de la HA, que es la glicoproteína de la superficie viral responsable de la unión al receptor de la célula huésped y la posterior fusión de membrana y entrada viral10.
La HA juega un papel importante en la respuesta inmune del huésped, estimulando la producción de anticuerpos neutralizantes responsables de la inmunidad protectora11-13.
Entre los virus de Paraguay estudiados algunos presentan mutaciones relacionadas a sitios antigénicos como K163Q, S185T, S203T y R205K. Los cambios de aminoácidos que ocurren en los sitios antigénicos o en la superficie de la molécula de HA pueden tener un efecto importante en el reconocimiento de anticuerpos, permitiendo al virus escapar de la respuesta inmune14. Entre otras mutaciones relevantes que presentan los virus estudiados, se encuentran las sustituciones de aminoácidos S185T dentro del sitio de unión al receptor, y I216V cerca de del mismo sitio, que pueden afectar la interacción de la HA con su receptor celular15.
Ninguno de los virus de Paraguay presento la mutación D222G que ha sido considerado un marcador de virulencia asociado con mayor severidad clínica16. Esta mutación ha sido asociada a un aumento de la avidez en la unión viral a receptores celulares que poseen ácido siálico unido a galactosa por un enlace α 2,317,8. Este tipo de receptores se encuentran en la región alveolar de los pulmones humanos lo que podría explicar la neumonía grave, al igual que en los casos humanos de infección H5N1, lo que estaría indicando la existencia de replicación viral en las vías respiratorias inferiores19. En concordancia con este hallazgo, los pacientes estudiados no manifestaron diferencias en la severidad clínica atribuibles a mutaciones virales, ya que los virus tanto de los casos ambulatorios como de los hospitalizados presentaron secuencias similares de aas dentro de un mismo periodo de tiempo. Por lo tanto habría que investigar otras causas para la severidad de la infección en algunos de estos pacientes, como comorbilidades o co-infecciones, lo cual escapa a los objetivos de este estudio.
Según estudios realizados en los centros colaboradores de OMS, los VI pertenecientes a todos los grupos genéticos detectados son antigénicamente indistinguibles del virus vacunal A/California/7/2009, a pesar de los cambios en aas y sitios de glicosilación20. Sin embargo según las recomendaciones para el hemisferio sur en el 2017, virus representativos del grupo emergente 6B.1, al cual pertenecen los virus de Paraguay aislados en el 2016, fueron poco inhibidos por algunos grupos de sueros humanos adultos posteriores a la vacunación. Además la media de los títulos de inhibición de sueros pediátricos pos vacunación contra algunos virus representativos del grupo 6B.1 se redujeron significativamente en comparación con los títulos contra el virus de la vacuna A/California/7/200921. Por ese motivo por primera vez desde la pandemia del año 2009, se sustituyó la cepa vacunal A/California/7/2009 por A/Michigan/45/2015. La nueva cepa vacunal pertenece al grupo 6B.1 caracterizado por la sustitución S162N que modifica un sitio de glicosilación, lo que podría conducir a cambios en el reconocimiento antigénico.
El análisis de la evolución genética del VI A(H1N1)pdm09 en Paraguay, indica que recién desde el año 2016 circularon en el país cepas pertenecientes al grupo antigénico emergente al cual pertenece la nueva cepa vacunal recomendada a partir del año 2017. Mientras que los virus circulantes en años anteriores pertenecen a grupos genéticos que resultaron antigénicamente similares a la cepa vacunal previa A/California/7/2009.
Conclusión
Estos resultados demuestran la importancia de la Vigilancia virológica sistemática y la caracterización genética de los virus de Influenza, en la provisión de datos oportunos para la formulación adecuada de vacunas recomendadas a nuestra región; así como en la detección de cambios en el genoma viral, que podrían estar relacionados a un aumento de la severidad clínica, de manera a alertar al sistema de salud para responder apropiadamente ante los brotes de influenza.
Reconocimientos
Agradecemos a CDC por el apoyo financiero para la adquisición de reactivos e insumos en el marco del Proyecto “Sostenibilidad de la Vigilancia de Influenza”; a OPS/OMS por el apoyo técnico para la Vigilancia virológica y epidemiológica de Influenza; al Fondo para la Convergencia Estructural de Mercosur (COF 03/11), por el apoyo en la contratación de las investigadoras María Liz Gamarra y Andrea Gómez de la Fuente.
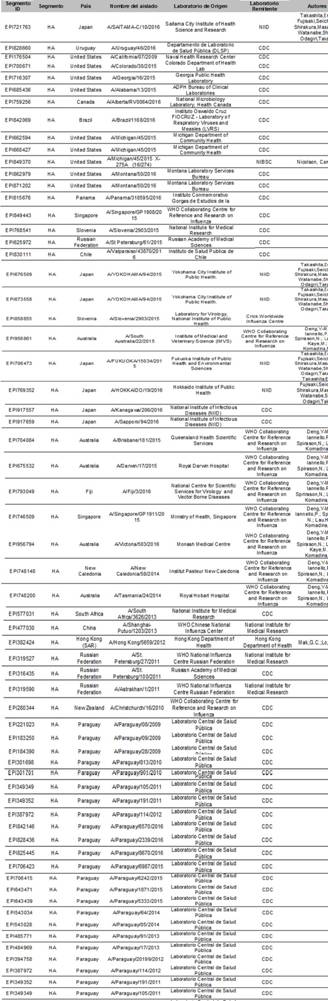
Reconocemos a los autores, laboratorios de origen y laboratorios que remitieron las secuencias disponibles en la base de datos GISAID’s EpiFlu™ en la cual esta investigación está basada. La lista se detalla a continuación